Enfermedades autoinmunes sistémicas más frecuentes en la adolescencia
A.L. Boteanu(1,2), L. Villalobos Sánchez(1,2)
(1)Hospital Universitario Ramón y Cajal. Madrid. (2)Clínica Universidad de Navarra. Madrid
Fecha de recepción: 14-01-2024
Fecha de publicación: 31 de marzo 2024
Adolescere 2024; XII (1): 27-43
Resumen
|
Las enfermedades autoinmunes sistémicas son un grupo heterogéneo de enfermedades, crónicas, potencialmente graves, que en un porcentaje bajo de casos puede debutar en la adolescencia. Dada su escasa prevalencia en la adolescencia, la heterogeneidad de los síntomas, muchas veces inespecíficos, suponen un desafío diagnóstico y terapéutico.
La mayoría tienen un predominio femenino. A lo largo de este artículo vamos a tratar temas como el lupus eritematoso sistémico (LES), el síndrome antifosfolipídico (SAF), la dermatomiositis (DM) y los síndromes escleridermiformes, con las peculiaridades que estas enfermedades pueden tener en cuanto al diagnóstico y manejo en la adolescencia. El periodo de transición entre la infancia y la edad adulta representa un periodo crucial en el desarrollo físico y psicosocial del individuo, lo que añade un grado de dificultad en cuanto al manejo de una enfermedad crónica compleja. El diagnóstico precoz es crucial para evitar daños y secuelas, igual que el inicio de un tratamiento adecuado. En la última década el desarrollo tecnológico ha llevado a un mejor conocimiento de las vías implicadas en las enfermedades y como consecuencia al desarrollo de una mayor número de fármacos. También se han publicado guías de práctica clínica y recomendaciones de tratamiento criterios, cada vez más haciendo hincapié en la importancia de un manejo personalizado. Tratándose de enfermedades crónicas, en ocasiones graves, el apoyo psicológico, aparte del tratamiento farmacológico, es fundamental para evitar complicaciones emocionales.
Palabras clave: Conectivopatias; Enfermedades autoinmunes sistémicas; Lupus eritematoso sistémico juvenil; Dermatomiositis juvenil; Síndrome antifosfolípido juvenil; Esclerodermia juvenil.
|
Abstract
|
Systemic autoimmune diseases are a heterogeneous group of chronic and potentially severe conditions, which, in a small percentage of cases can start in adolescence. Given their low prevalence in adolescence and the heterogeneity of the symptoms, which are often nonspecific, the diagnosis can be challenging and is often delayed. Most of the systemic autoimmune diseases have a female predominance. Throughout this article, topics such as systemic lupus erythematosus (SLE), antiphospholipid syndrome (APS), dermatomyositis (DM), and scleroderma-like syndromes will be discussed, focusing on the peculiarities these diseases may have in terms of diagnosis and management in adolescence. The transition period between childhood and adulthood represents a crucial time in an individual's physical and psychosocial development, adding a level of difficulty in managing a complex chronic illness. Early diagnosis is crucial to prevent damage, just like the initiation of appropriate treatment. Over the last decade, technological developments have led to a better understanding of the pathways involved in these diseases and, as a consequence, the development of a greater number of drugs. Clinical practice guidelines and treatment recommendations have also been published, increasingly emphasizing the importance of personalized management. Given that these are chronic and sometimes severe diseases, psychological support, in addition to pharmacological treatment, is essential to prevent emotional complications.
Key words: Connective tissue diseases; Systemic autoimmune diseases; Juvenile systemic lupus erythematosus; Juvenile dermatomyositis; Juvenile antiphospholipid syndrome; Juvenile scleroderma.
|
LUPUS ERITEMATOSO SISTÉMICO (LES)
Introducción
En niños y adolescentes la gravedad de los síntomas de LES suele ser mayor que en adultos
El LES es una enfermedad autoinmune en la que se puede afectar cualquier órgano o sistema y se caracteriza por la presencia de autoanticuerpos. La clínica es variable en cuanto a asociación de síntomas, en cuanto al curso evolutivo o a la gravedad, siendo una enfermedad potencialmente mortal. En niños y adolescentes la gravedad de los síntomas suele ser mayor que en adultos. Esto unido al retraso en el diagnóstico y la ausencia de estudios específicamente diseñados para la edad pediátrica que valoren la eficacia y los efectos secundarios posibles, hace que haya más daño a corto y largo plazo que en el lupus de debut en adultos(1).
Epidemiología
El 15-20 % de los LES debutan en la edad infantil con un pico en la adolescencia entre los 12-14 años
Un 15-20 % de los LES debutan en edad infantil con un pico del debut en la adolescencia, entre los 12 y los 14 años. Se calcula que la incidencia del LES pediátrico (LESp) es de 0,36 – 2,5/ 100.000 niños. La prevalencia estimada es de 1,89 –34,1/100.000 niños. La edad media de presentación es de 11-12 años, siendo infrecuente antes de los 5 años. En los niños con debut precoz (< 5 años) la distribución por géneros (mujeres: varones = 1:1) no muestra la diferencia que se observa en la edad prepuberal (7-13 años) cuando la ratio mujeres: varones es de 4-5:1 o después de la pubertad (13-18 años) cuando la ratio es similar al observado en adultos (mujeres: varones = 9-10:1). La enfermedad es más frecuente en niñas de ascendencia africana, asiática o hispana y menos frecuente en europeos, con una importante diferencia de la prevalencia entre los continentes.
Etiopatología
Similar a otras enfermedades autoinmunes, la etiopatogenia es desconocida, multifactorial en la mayoría de los pacientes. Existen varias teorías, ninguna de ellas explica en totalidad la patogenia de la enfermedad en todos los pacientes. Se reconoce la implicación de factores genéticos, epigenéticos, ambientales exógenos y/o endógenos e inmunológicos.
Cuando la enfermedad debuta en la infancia, los factores genéticos tienen una mayor contribución que en los adultos. En un pequeño porcentaje de pacientes (1-3 %), sobre todo los que debutan en edades muy tempranas, se ha demostrado la presencia de mutaciones en genes relacionados de forma directa o indirecta con la vía del interferón (LES monogénico). Además, estudios genéticos recientes han identificado más de 100 genes o variantes genéticas asociadas a un incremento en el riesgo para desarrollar la enfermedad(2).
Los factores ambientales como la exposición a la luz ultravioleta, algunos virus (VEB, CM, etc.), o exposición a fármacos pueden desencadenar un LES en niños predispuestos genéticamente. La diferencia en la incidencia de la enfermedad en la vida fértil de la mujer sugiere que las hormonas sexuales femeninas juegan un importante papel en la etiopatogenia de la enfermedad.
La rotura de la tolerancia inmune y la desregulación del sistema inmune lleva a la producción de autoanticuerpos (autoAc), especialmente antinucleares (ANA) que son una constante en los pacientes con LES (> 90 % adolescentes con LES tienen ANA positivos). La presencia de estos autoanticuerpos (ANA, anticuerpos anti-DNA, anticuerpos anti -Sm, etc.) son responsables de parte de la inflamación y el daño mediante la formación de inmunocomplejos. En estudios recientes se ha observado que los autoanticuerpos son más frecuentemente presentes en niños con LES, comparando con el debut en la edad adulta.
Manifestaciones clínicas
Las manifestaciones clínicas del LES en niños y adolescentes son similares a las observadas en adultos. Hay una importante variabilidad en cuanto a la gravedad entre los pacientes. Varios estudios han puesto de manifiesto que las manifestaciones clínicas que implican la afectación de un órgano mayor (sobre todo renal o neurológico) son más frecuentes en niños que en adultos y de mayor gravedad. Un porcentaje importante de pacientes van a presentar manifestaciones graves al debut de la enfermedad. La clínica incluye síntomas sistémicos inespecíficos como la fiebre, astenia, o pérdida de peso y/o síntomas específicos para varios sistemas y órganos.
Las manifestaciones musculoesque-léticas del LES son frecuentes y en muchas ocasiones los síntomas del debut
Las manifestaciones musculoesqueléticas (artralgias de ritmo inflamatorio y mialgias) son frecuentes y en muchas ocasiones uno de los síntomas de debut. Dada la intensa actividad física habitual de los adolescentes, las artralgias se interpretan como secundarias a la actividad física. Una adecuada anamnesis donde destaca el carácter inflamatorio del dolor (mejora con la actividad física, se desencadena con el reposo y después del descanso nocturno) puede aumentar la sospecha diagnóstica. La artritis se presenta en un porcentaje que varía entre el 20 y el 74 % de los pacientes, según la serie publicada. Afecta a grandes y pequeñas articulaciones y no suele ser erosiva. Puede ser transitoria y autolimitada. La tenosinovitis, sobre todo de las manos, puede ser ocasional y transitoria o puede llevar a la aparición de roturas y deformidades.
Las manifestaciones mucocutáneas son frecuentes en el LESp. Pueden ser muy variadas, pero suelen repetir el mismo patrón en un paciente. Dado que son muy variadas existe una cierta dificultad en su clasificación. El rash malar (Figura 1), el rash discoide, las ulceras orales (Figura 2) y la fotosensibilidad son las manifestaciones incluidas en los criterios de clasificación ACR 1997. En los criterios EULAR/ACR 2019 se han incluido la alopecia no cicatricial (2p) úlceras orales (2p), lupus cutáneo subagudo o discoide (4p), lupus cutáneo agudo (6p) mientras que los criterios SLICC 2012 incluyen un número mayor de lesiones. El rash malar y las ulceras orales son más frecuentes en niños que en los adultos. Algunas de las lesiones son específicas (con dermatitis de interfase en la microscopia) como el lupus cutáneo agudo, subagudo o crónico, pero también se pueden observar lesiones inespecíficas (vasculitis, vasculopatía, ampollosas, perniosis, etc.).
Las manifestaciones renales y neurológicas del LES son graves y más frecuentes en niños que en adultos
La afectación renal y neurológica son manifestaciones graves, más frecuentes en niños que en adultos. La nefritis lúpica afecta al 60-80 % de los pacientes. Las formas de presentación incluyen: proteinuria, hematuria microscópica, síndrome nefrótico, hipertensión arterial o deterioro de la función renal. Se recomienda realizar biopsia renal en los pacientes con afectación renal. La nefritis lúpica diagnosticada mediante biopsia renal se clasifica según el sistema de clasificación International Society of Nephrology/Renal Pathology Society (ISN/RPS) 2003 en 6 clases, siendo las clases III y IV las más frecuentemente observadas en adolescentes. Entre las manifestaciones neuropsiquiátricas más importantes se encuentran las crisis epilépticas, siendo más frecuentemente observadas en niños que en los adultos. Otras manifestaciones neuropsiquiátricas incluyen: cefalea, trastorno del ánimo, enfermedad cerebro vascular, corea, meningitis aséptica y neuropatía de nervios craneales o periféricos. Los cuadros neuropsiquiátricos son motivo de un diagnóstico diferencial exhaustivo y en ocasiones complejo (infecciones, efecto secundario a fármacos, etc.)
Manifestaciones cardiorrespiratorias. La pleuro-pericarditis es la manifestación cardiorrespiratoria más frecuentemente encontrada en los pacientes con LESp (30 %). Otras manifestaciones: miocarditis, endocarditis aséptica, enfermedad pulmonar intersticial, hemorragia alveolar, neumonitis, pulmón encogido. La afectación de arterias coronarias es muy rara en niños.
Las manifestaciones gastrointestinales son relativamente frecuentes en el LESp. siendo los más frecuentes el dolor abdominal (inespecífico o secundario a serositis o pancreatitis, etc.). Otras manifestaciones gastrointestinales son: hepatoesplenomegalia, hepatitis autoinmune, colitis, disfunción esofágica, etc.
Manifestaciones hematológicas e inmunológicas. La anemia se encuentra en más del 50 % de los pacientes; se caracteriza por ser una anemia de trastornos crónicos en la mayoría de los casos. Es frecuente la presencia de un test de Coombs directo (TCD) positivo sin signos de hemólisis. Ocasionalmente se presenta anemia hemolítica (TCD positivo, sideremia, reticulocitos y bilirrubina no conjugada aumentadas con disminución de haptoglobina) o más raramente anemia hemolítica microangiopática (TCD negativo, trombopenia, aumento de dímeros D, esquistocitos en sangre periférica y en ocasiones alteración renal y/o neurológica). La leucopenia (< 4.000/mm3), y/o la linfopenia (<1.500/mm3) o la trombocitopenia (< 100.000/mm3) son frecuentes y pueden persistir durante períodos prolongados incluso en ausencia de actividad inflamatoria. Otras alteraciones en las determinaciones bioquímicas o de la coagulación son inespecíficas o suelen ser órganos dependientes (deterioro de la función renal, hepática, afectación renal lúpica, etc.).
Excepto al Ac anti-DNA nativo que se relaciona con la actividad del LESp, la positividad de los restantes autoAC no constituyen un buen índice de actividad de la enfermedad
Alteraciones inmunológicas. Autoanticuerpos: En más de 90 % de los niños con LES se detectan anticuerpos antinucleares (ANA) y en el 54-93 % anticuerpos anti-DNA. Con la excepción de los anticuerpos anti-DNA nativo, la positividad de los restantes autoAC, no constituye un buen indicador de la actividad de la enfermedad, por lo cual se recomienda su determinación para fines de diagnóstico, pero no para la monitorización de la actividad. Por el contrario, los anticuerpos anti-DNA se han relacionado con la actividad del LESp y se incluyen en los índices de actividad. Los autoanticuerpos se pueden observar en un porcentaje de pacientes asintomáticos y sin una enfermedad autoinmune presente. De los más de 120 autoAc observados en el LES, los más relevantes hasta la fecha han sido los ANA, anti-DNAds, anti-Sm, anti-RNP, anti- Ro/SS-A y anti-La/SS-B, sin embargo, no se ha demostrado especificidad predictiva para ninguno de ellos. Ocasionalmente existe positividad para los anticuerpos antifosfolípidos, asociados o no a un SAF.
Los anticuerpos ANCA suelen ser negativos y se utilizan para el diagnóstico diferencial. Complemento: en los pacientes con LESp se puede observar un déficit del complemento por consumo, actividad o deficiencias genéticas del mismo. Los niveles disminuidos de CH50, C3 y C4 se asocian con la actividad clínica. El descenso de C4 suele preceder los brotes, aunque en ocasiones puede no estar en relación con actividad clínica.
Diagnóstico
No existe una única prueba para el diagnóstico del LESp
No existe una única prueba específica para el diagnóstico de LESp. El diagnóstico suele ser difícil dada la inespecificidad de la mayoría de los síntomas, siendo necesario un amplio diagnóstico diferencial. Los anticuerpos ANA, que en ocasiones se usan como screening, se pueden detectar tanto en población sana como en otras enfermedades autoinmunes (artritis idiopática juvenil, dermatomiositis, esclerodermia, Síndrome Sjögren, etc.). Hasta la fecha no se han diseñado criterios de clasificación específicos para los niños y adolescentes con LES, usándose los propuestos para adultos:
- Los criterios de clasificación establecidos por el Colegio Americano de Reumatología (ACR) en 1982 y revisados en 1997, precisando para el diagnóstico 4 de los 11 items(3).
- Los criterios SLICC (Systemic Lupus International Collaborating Clinics) publicados en 2012, proponen cambios importantes, incluyendo un mayor número de manifestaciones clínicas y analíticas. Se requiere la presencia de 4 criterios de los cuales al menos 1 debe ser clínico y 1 inmunológico o presentar nefritis lúpica (demostrada mediante biopsia) en presencia de ANA o de anti-DNAd(4).
- Los criterios EULAR/ACR publicados en 2019(5), que incluyen manifestaciones clínicas y analíticas, cada una de ellas con una puntuación diferente, partiendo de un criterio de entrada: ANA positivos a título ≥ 1/80 realizado en células HEp2 o equivalente. Los criterios no necesitan cumplirse simultáneamente. En cada categoría solo se cuenta el criterio con la puntuación más alta. Para cumplir los criterios de clasificación de LES se requiere ≥ 1 criterio clínico y ≥ 10 puntos.
Un estudio recientemente publicado pone de manifiesto que los criterios EULAR/ACR publicados en 2019 son los que mejor clasifican los pacientes adultos, sin embargo, en población pediátrica son los criterios SLICC 2012 los más recomendados para utilizar(6).
Tratamiento
El tratamiento del LES será integral y personalizado: controlar la actividad de la enfermedad, disminuir el daño por la enfermedad y uso de fármacos, mejorar la calidad de vida y el impacto emocional
El LES en la infancia y adolescencia puede impactar significativamente el crecimiento y el desarrollo, así como la calidad de vida, aparte de producir daños orgánicos importantes, lo que subraya la importancia de un enfoque integral que aborde tanto los aspectos físicos como psicosociales de la enfermedad. El tratamiento es complejo y debe de ser personalizado, centrado en controlar la actividad de la enfermedad, disminuir el daño producido tanto por la enfermedad como por los fármacos recibidos y mejorar la calidad de vida y el impacto emocional. Las estrategias terapéuticas van a depender del órgano o sistema afectado. Los pacientes pueden requerir altas dosis de corticoides y fármacos inmunosupresores debido a la mayor actividad en esta edad.
Los pilares de tratamiento se basan en medidas generales, no farmacológicas (evitar la exposición solar, aplicación de cremas de fotoprotección (IPS < 50), inmunización según el calendario vacunal, al ser posible previo al inicio de la terapia inmunosupresora, etc.). El tratamiento convencional se basa en corticoides, fármacos modificadores de la enfermedad (FAME) convencionales (Hidroxicloroquina, Azatioprina, Micofenolato, Metotrexato, ciclofosfamida, anticalcineurínicos, etc.) o FAME biológicos (Belimumab, Rituximab, etc). La afectación de órgano mayor (renal, neurológica, cardiaca), precisan habitualmente un tratamiento inmunosupresor intensivo para controlar la clínica (ciclofosfamida, micofenolato, rituximab, belimumab). El número y la calidad de los estudios específicamente diseñados para la población pediátrica es escasa, por lo tanto, las recomendaciones se basan en la experiencia en adultos y muchas veces en estudios observacionales y series de casos. El único fármaco que dispone de ensayos clínicos y está aprobado por las agencias reguladoras para su uso en los niños con LES es el Belimumab (anticuerpo monoclonal que inhibe el factor estimulador del linfocito B – BLyS). Otros fármacos como en el Obinutuzumab (anticuerpo antiCD-20) o el Anifrolumab (anticuerpo anti receptor del interferón tipo I) están actualmente en fase de investigación. Además, la atención integral debe incluir asesoramiento nutricional, apoyo psicológico y educación sobre la enfermedad, fundamental para asegurar una buena adherencia al tratamiento, ayudar a los adolescentes a manejar su enfermedad y afrontar los desafíos físicos y emocionales asociados.
El único fármaco que dispone de ensayos clínicos y está aprobado por las agencias reguladoras para su uso en niños con LES es el Belimumab
SÍNDROME ANTIFOSFOLÍPIDO (SAF)
Introducción
El SAF consiste en la presencia de Ac antifosfolípidos que incrementan el riesgo de trombosis y complicaciones obstétricas. Puede ser aislada o asociada a otras enfermedades autoinmunes como LES
El síndrome antifosfolípido en niños y adolescentes es una enfermedad rara y compleja, de difícil diagnóstico ante la ausencia del criterio obstétrico, que requiere un enfoque personalizado para su manejo. Es una de las trombofilias adquiridas más frecuentes. Se caracteriza por la presencia de anticuerpos antifosfolípidos (aPL) que incrementan el riesgo de trombosis y complicaciones obstétricas. Puede presentarse como una enfermedad aislada (síndrome antifosfolípido primario) o asociada a otras enfermedades autoinmunes como el LES (SAF secundario). Un diagnóstico temprano, tratamiento efectivo, así como la educación, son esenciales para mejorar los resultados y la calidad de vida de los adolescentes con SAF.
Manifestaciones clínicas y analíticas
En ausencia de componente obstétrico (infrecuente en adolescentes), las manifestaciones clínicas más características son las trombosis venosas o arteriales y complicaciones de las mismas. Las trombosis pueden ocurrir en cualquier vaso, pero es más frecuente que se presente en las venas de los miembros inferiores (60 %), seguido de trombosis arterial (30 %) y trombosis de pequeño vaso(7). Las trombosis son menos frecuentes en niños que en adultos, sin embargo, el número de trombosis que se atribuyen al SAF es mayor que en población adulta, dada la ausencia de otros factores de riesgo cardiovasculares o de otra naturaleza que lo justifique. Las manifestaciones no-criterio más frecuentes en población pediátrica son las hematológicas (trombocitopenia autoinmune, anemia hemolítica autoinmune, Sd. Evans). Otras manifestaciones clínicas: livedo reticularis/racemosa, síndrome Raynaud, endocarditis no infecciosa, amaurosis, cefalea, epilepsia, corea, Sd. Budd-Chiari, trombosis de venas portales, etc. Las manifestaciones clínicas consideradas “criterio no trombótico” son frecuentes en población pediátrica.
Las recomendaciones SHARE sugieren que el perfil de anticuerpos aPL, incluyendo anticoagulante lúpico (ALE), anticardiolipinas (aCL) IgG e IgM, y anti-β2GPI IgG e IgM, debe ser considerado para todos los niños y adolescentes con sospecha de trombofilia autoinmune(8). Otro de los retos del SAF en población pediátrica es la presencia de anticuerpos aPL transitorios relacionados con infecciones y vacunas. Por lo tanto, se necesitan títulos altos y positividad persistente de aPL para cumplir con los criterios de laboratorio. Además estos deben de evaluarse en dos o más ocasiones con al menos 12 semanas de diferencia, especialmente en niños, y adolescentes.
Las pruebas positivas para ALE están asociadas con un mayor riesgo de trombosis mientras que los anticuerpos aCL y ALE han demostrado más sensibilidad y especificidad, respectivamente.
Diagnóstico
Igual que ocurre en otras enfermedades raras, no existen unos criterios diagnósticos o de clasificación especialmente diseñados para su uso en población pediátrica, lo que en muchas ocasiones conlleva un retraso en el diagnóstico. Hasta el año 2023 la clasificación del SAF se realizaba en base a los criterios de Sapporo de 1999, revisadas en 2006(9), que consisten en morbilidad obstétrica o presencia de trombos arteriales o venosos y la presencia de anticuerpos aPL (2 determinaciones positivas separadas al menos 12 semanas). Dado el aumento del conocimiento de esta enfermedad y la colaboración internacional, en 2023 se propusieron unos nuevos criterios de clasificación, que muestran una mayor sensibilidad y especificidad en adultos, comparando con los anteriores. Estos criterios que se han desarrollado conjuntamente por el Colegio Americano de Reumatología (ACR) y EULAR, introducen un enfoque más detallado y específico para la clasificación de esta enfermedad, incluyendo con una ponderación variable, varios síntomas o signos “no- criterio” en la clasificación de Sapporo. Los nuevos criterios incluyen un criterio de entrada de al menos una prueba positiva de anticuerpos antifosfolípidos (aPL) dentro de los 3 años posteriores a la identificación de un criterio clínico asociado a aPL, seguido de criterios ponderados clínicos y analíticos (Tabla I)(10). Está pendiente su validación en la población pediátrica.
Tratamiento
El objetivo principal del tratamiento del SAF es la profilaxis primaria o secundaria de la trombosis
El objetivo principal es la profilaxis primaria o secundaria de la trombosis. Las estrategias de manejo en adolescentes deben incluir la evaluación de factores de riesgo protrombóticos adicionales (ej. consumo de tabaco, obesidad, dislipemia, etc.) y la consideración de estrategias anticonceptivas (evitar anticonceptivos con estrógenos en adolescentes en edad fértil), recomendaciones sobre el estilo de vida (el consumo de alcohol puede interferir con el tratamiento con Sintrom, evitar deportes de contacto ante el aumento de riesgo de hemorragia en pacientes anticoagulados, etc.), adherencia al tratamiento y apoyo psicológico.
En niños y adolescentes, se ha propuesto una dosis inicial de heparina de bajo peso molecular (HBPM) de enoxaparina de 1,0 mg/kg subcutáneo cada 12 horas. En caso de trombosis asociada con positividad persistente de aPL, se debe indicar anticoagulación a largo plazo. Los antagonistas de la vitamina K (ej.: Sintrom) son el tratamiento estándar, siendo el objetivo de INR de 2,0–3,0 para eventos venosos. Para la trombosis recurrente, se debe evaluar sistemáticamente la adherencia al tratamiento y otros factores de riesgo asociados. En este caso se podría considerar un nuevo objetivo de INR de 3,0–4,0 o terapias alternativas, como una dosis terapéutica extendida de HBPM. Las recomendaciones EULAR también sugieren agregar aspirina en baja dosis con los antagonistas de la vitamina K, como alternativa a un INR más alto. En caso de afectación arterial, también se indica la combinación de agentes antiplaquetarios y anticoagulación, y el objetivo de INR podría ser de 3,0–4,0. La evidencia para la trombosis venosa recurrente a pesar del tratamiento adecuado es limitada. Se debe considerar también el uso de hidroxicloroquina como terapia adyuvante para el APS. Aunque el uso de anticoagulantes orales directos podría representar un potencial avance para el tratamiento por la facilidad de la administración sin requerir controles analíticos frecuentes, las sociedades científicas recomiendan evitar estrictamente su uso en pacientes con síndrome antifosfolípido (APS) con trombosis arterial, de pequeños vasos y recurrente, o enfermedad valvular cardíaca, y en aquellos con triple positividad(11).
Los glucocorticoides intravenosos en pulsos, la plasmaféresis, la administración de inmunoglobulinas, el Rituximab o el Eculizumab son estrategias posibles en caso de síndrome antifosfolipídico catastrófico, aunque la experiencia es muy limitada en población pediátrica.
DERMATOMIOSITIS JUVENIL (DMI)
Introducción
La dermatomiositis juvenil (DMJ) es una enfermedad autoinmune sistémica que se caracteriza por afectación cutánea (principalmente pápulas de Gottron y rash heliotropo) y debilidad muscular de inicio insidioso(12). Dada la afectación multisistémica es importante un manejo multidisciplinar entre pediatras, dermatólogos y reumatólogos expertos en patología autoinmune infantil en centros de referencia.
Epidemiología
A pesar de su baja incidencia (2-4/1.000.000/año en la edad pediátrica) es la miopatía más frecuente en la infancia(13,14). Su presentación es más frecuente en mujeres, y el debut principalmente entre los 5 y los 14 años(12).
Fisiopatología
La fisiopatología es aún desconocida, pero la combinación de predisposición genética junto a factores ambientales (infecciones, rayos ultravioletas, fármacos) parecen estar en la base de la enfermedad, como en otras enfermedades autoinmunes sistémicas(15). A diferencia de lo observado en adultos, en niños la asociación con la neoplasia es excepcional, por lo tanto, no se incluye en el diagnóstico diferencial de estas de forma rutinaria.
Manifestaciones clínicas
Las manifestaciones cutáneas y osteomusculares de la DMI son las más características. Con adecuado y precoz tratamiento, el 30-50 % consiguen remisión en 2-3 años
A nivel clínico lo más característico son las manifestaciones cutáneas, como rash heliotropo, pápulas de Gottron (Figura 3), eritema periungueal, úlceras cutáneas y el signo del chal. Es frecuente la calcinosis en la DMJ en comparación con la DM de presentación en adulto.
A nivel osteomuscular los pacientes presentan mialgias y debilidad insidiosa, de predominio proximal, con dificultad para levantarse del suelo. Pueden asociar artritis de rodillas, carpos o pequeñas articulaciones de las manos, principalmente (40-60 %), y contracturas musculares(16).
Pueden presentar así mismo otras manifestaciones como fiebre, fenómeno de Raynaud, lipodistrofia (8-40 %), calcinosis (30-70 %), enfermedad pulmonar intersticial (8 %), disfagia y, en raras ocasiones, manifestaciones cardiacas (pericarditis, endocarditis, arritmias)(12,14,16).
El curso de la enfermedad es variable, entre monofásico y curso crónico (41-60 %), con periodos intermitentes de actividad-remisión. Con un adecuado y precoz tratamiento, el 30-50 % de los pacientes consiguen remisión en 2-3 años con tasas de mortalidad debajo del 4 %(13,14).
Las secuelas potenciales incluyen contracturas musculares, calcinosis subcutánea y discapacidad funcional(16).
Diagnóstico
El diagnóstico se establece según criterios clínicos, analíticos y de pruebas complementarias. Para la DMJ utilizamos los criterios de clasificación EULAR/ACR (Tabla II)(13).
A nivel analítico las enzimas musculares suelen estar elevadas (principalmente en DMJ miopática, entre 87-100 % presentan elevación), puede haber elevación de reactantes de fase aguda, linfopenia y alteración del perfil hepático.
Los ANA son positivos en hasta el 85 % de los casos. La positividad de anticuerpos específicos se presenta en torno al 60 % de los pacientes con DMJ (Tabla III, diferencias entre patrón miopático y amiopático y características fenotípicas asociadas)(13).
La RNM muscular es útil al debut de la DMI para valorar la inflamación muscular y guiar la biopsia muscular
La RNM muscular es una herramienta útil al debut para valorar inflamación muscular y durante el seguimiento para diferenciar actividad/inactividad. Es útil también para guiar una biopsia muscular, la cual se recomienda en casos atípicos o con dudas diagnósticas. El EMG nos valoran datos de afectación muscular inflamatoria y puede ser útil en casos dudosos y debe ser realizado por expertos neurofisiólogos(16).
Según las manifestaciones sistémicas, se realizarán las pruebas complementarias oportunas (capilaroscopia en caso de Raynaud, estudio radiológico torácico en caso de posible afectación pulmonar, radiografías simples para valorar calcinosis…)(2). De manera general se aconseja realizar al inicio radiografía de tórax, pruebas de función respiratoria incluyendo capacidad de difusión de monóxido de carbono, ECG y ecocardiograma, ya que las manifestaciones cardiopulmonares pueden cursar asintomáticas y pueden ser posibles causas de mortalidad(14).
Diagnóstico diferencial
El diagnóstico diferencial debe incluir: distrofias musculares, miopatías metabólicas, atrofia muscular espinal, enfermedades de motoneurona, miastenia gravis, otras enfermedades autoinmunes sistémicas, enfermedades endocrinológicas hipo y hipertiroidismo, hipo y hipercalcemia, e hipopotasemia), miopatías farmacológicas y miopatías infecciosas(13,16).
Tratamiento
El objetivo incluye control de la actividad de la enfermedad, evitar daño orgánico y mejorar la calidad de vida(14).
Medidas no farmacológicas
Incluye la fotoprotección solar, terapia física rehabilitadora, y en algunos casos, terapia psicológica si se precisa, para el paciente y/o familiares. Hay que asegurarse, como en otras conectivopatías, del correcto calendario vacunal y realizar tratamiento precoz de las infecciones.
Medidas farmacológicas
Según las recomendaciones CARRA (Childhood Arthritis and Rheumatology Research Alliance) y SHARE (Single Hub and Acces point for the pediatric Rheumatology in Europe) el tratamiento debe individualizarse y adaptarse a la severidad de la presentación. El tratamiento inicial son los corticoides (bolos de metilprednisolona durante 3 días, seguidos de prednisona a dosis de 1-2mg/Kg/día con descenso progresivo) combinados un fármaco modificador de la enfermedad, como la hidroxicloroquina y/o metotrexato (dosis de 15-20 mg/m2/semana). Para los casos refractarios se podría valorar tratamiento con inmunoglobulinas, ciclofosfamida, micofenolato, azatioprina, fármacos anti TNF (infliximab, adalimumab), inhibidores JAK o rituximab según las manifestaciones clínicas y la gravedad de presentación. Durante el tratamiento con corticoides se debe añadir calcio/vitamina D para prevenir fracturas y minimizar la osteoporosis y considerar septrim en casos de corticoterapia prolongada a altas dosis(16).
El tratamiento inicial de la DMI son los corticoides combinados con un fármaco modificador de la enfermedad (hidroxicloroquina y/o metotrexato)
La dermatomiositis juvenil, por la tanto, se trata de una enfermedad autoinmune que potencialmente puede producir afectación multisistémica, presentándose principalmente con manifestaciones cutáneas y musculares. Es importante conocerla bien para sospecharla y derivarla a centros de referencia, ya que el tratamiento precoz mejora el pronóstico.
ESCLEROSIS SISTÉMICA JUVENIL
Introducción
La esclerosis sistémica juvenil es una enfermedad autoinmune sistémica rara caracterizada por inflamación, vasculopatía y fibrosis con afectación cutánea y multiorgánica(17).
Epidemiología
Se estima una prevalencia de 3 cada 1.000.000 niños(18). Predomina en sexo femenino, raza caucásica y la edad media de presentación es 9 años(19).
Clínica
El fenómeno de Raynaud es una de las características más frecuentes (70-90 %), siendo con frecuencia el primer síntoma (incluso puede preceder en años a la enfermedad) y puede conducir, en casos severos, a úlceras digitales.
El primer síntoma y el más frecuente de la esclerosis sistémica juvenil es el fenómeno de Raynaud. La segunda manifestación es el endurecimiento cutáneo, la forma difusa afecta a tronco y extremidades y la limitada afecta a extremidades, cabeza y cuello
El endurecimiento cutáneo es la segunda manifestación más frecuente en el curso de la enfermedad y está presente en casi la totalidad de los pacientes (solo un pequeño porcentaje presentan esclerosis sistémica sin esclerodermia). La afectación cutánea facial da lugar a la facie típica: apertura oral limitada, microstomia, nariz afilada, labios delgados. La extensión de la afectación cutánea nos lleva a clasificarla en: esclerosis sistémica cutánea difusa (ESCD) que afecta tronco y regiones proximales de extremidades, y esclerosis sistémica cutánea limitada (ESCL), que afecta a regiones distales de extremidades, cabeza y cuello. La cuantificación de afectación cutánea es un indicador de severidad y un marcador de respuesta a tratamiento.
Las manifestaciones músculo-esqueléticas son más comunes que en adultos (60 %): artritis, fibrosis de tendones, contracturas musculares, miopatía con debilidad muscular y atrofia y/o miositis (con más riesgo de afectación cardiaca).
La afectación multiorgánica incluye al sistema pulmonar, cardiaco, gastrointestinal, renal y sistema nervioso, siendo más frecuentes los dos primeros.
La afectación gastrointestinal es muy frecuente (más del 70 % de los casos) con afectación principalmente esofágica manifestándose como disfagia. El reflujo gastroesofágico es otra de las manifestaciones que puede llegar a ocasionar esófago de Barret. Otras manifestaciones incluyen plenitud postprandial, saciedad precoz, distensión abdominal, meteorismo, pérdida de peso, estreñimiento e incontinencia anal, entre otras.
La afectación pulmonar ocurre en 35-55 %, incluyendo enfermedad pulmonar intersticial e hipertensión pulmonar (<10 %). Puede cursar asintomática, luego es importantísimo el screening.
La afectación cardiaca es rara (5-15 %), pero es la principal causa de mortalidad.
La afectación renal es inferior al 5 %. Las crisis renales son muy raras en niños.
A nivel de sistema nervioso se pueden presentar neuropatías periféricas y craneales.
Diagnóstico
La esclerodermia juvenil no suele ser bien identificada lo que conduce a un retraso diagnóstico en la mayoría de los casos, por lo tanto, es importante conocer la enfermedad para una derivación temprana a los pacientes con fenómeno de Raynaud muy precoz o severo, edema de manos, positividad de anticuerpos o manifestaciones cutáneas esclerodermiformes.
Los pacientes con alteraciones capilaroscópicas, Raynaud y ANA positivos se clasifican como pre-esclerodermia
Todo niño derivado a la consulta por Raynaud se le debe solicitar estudio inmunológico y capilaroscopia. La capilaroscopia es una técnica no invasiva para valoración vascular en la que debemos buscar como patrón de esclerodermia, áreas avasculares, dilataciones, ramificaciones y alteración de la arquitectura. Los pacientes con alteraciones capilaroscópicas, Raynaud y ANA positivos se clasifican como pre-esclerodermia(19).
Para el diagnóstico nos basamos en los criterios de clasificación PRES/ACR/EULAR que se basa en un criterio mayor (afectación cutánea) y criterios menores (esclerodactilia, afectación vascular, gastrointestinal, renal, cardiaca, respiratoria, musculoesquelética, neurológica o criterios serológicos).
Como pruebas complementarias para diagnóstico y screening de posible afectación orgánica utilizamos de manera sistemática capilaroscopia, ecocardiograma, electrocardiograma, pruebas de función respiratoria, test de la marcha de 6 minutos, TAC de tórax y analítica. Se debe valorar siempre el estado nutricional del paciente y el adecuado crecimiento. En el caso de síntomas gastrointestinal se solicitará estudio dirigido (manometría, pHmetría…).
A nivel analítico no suele haber alteraciones específicas. En el perfil de autoinmunidad, los ANA son positivos en prácticamente la mayoría de los casos.
Diagnóstico diferencial
El diagnóstico diferencial siempre va a incluir otras conectivopatías, pero también tenemos que diferenciar la ES de las esclerodermias localizadas y de otros síndromes esclerodermiformes.
a) Esclerodermia localizada o morfeas
Según la clasificación propuesta por PReS, las morfeas se dividen en 5 tipos: morfea lineal o en banda (la forma más frecuente en niños, en 51-65 %), la morfea circunscrita (superficial y profunda), morfea mixta, morfea generalizada y morfea panesclerótica. Dentro de las morfeas lineales hay 3 subtipos:
- Morfea en banda
- Morfea en coup de sabre, afectando a la frente (desde cuero cabelludo a la ceja)
- Sindrome Parry Romberg o hemiatrofia facial progresiva
Podemos observar alteraciones analíticas (como elevación de reactantes de fase aguda en formas profundas, elevación de glóbulos blancos, eosinofilia, elevación de creatin kinasa…) pero no hay correlación con la actividad de la morfea ni con el pronóstico(20).
b) Sindromes esclerodermiformes
Fascitis eosinofílica
Su presentación es rara en adolescentes. Se caracteriza por induración cutánea y tejidos subcutáneos y eosinofilia. Predomina en manos y tobillos y hay mayor afectación muscular que en adultos. El tratamiento es con corticoides y metotrexato con mejor pronóstico.
Enfermedad injerto contra huésped
Se produce afectación cutánea que puede llegar a ser simultánea junto con manifestación articular y contracturas. Se trata con corticoides, tacrolimus, metotrexato y/o micofenolato(21).
Tratamiento
El tratamiento de la esclerosis sistémica juvenil debe evaluarse con un equipo multidisciplinar. Se realizará valoración cutánea y orgánica y empleo de fármacos sistémicos (corticoides, metotrexato o micofenolato)
El tratamiento irá dirigido a las diferentes manifestaciones sistémicas y cutáneas.
El grupo de trabajo SHARE ha formulado 14 recomendaciones para el manejo de la ES juvenil.
De manera general recomiendan valoración cutánea y orgánica y manejo con fármacos sistémicos. Es importante el manejo por un equipo multidisciplinar de expertos en la enfermedad.
Los corticoides están indicados al inicio, especialmente en casos de miositis o artritis, sin haber un consenso en dosis y régimen de administración. Metotrexato (dosis 15mg/m/semanal) u otro fármaco modificador de la enfermedad también está indicado al inicio principalmente en casos de afectación cutánea, articular, vascular y/o gastrointestinal. Si el metotrexato no es eficaz se recomienda cambiar o añadir micofenolato.
Para la afectación intersticial pulmonar y /o cardiaca se recomienda ciclofosfamida, con precaución en su uso.
Para la afectación vascular se usan los inhibidores de los canales de calcio, inhibidores de la fosfodiesterasa 5 y/o análogos de la prostaciclina. Bosentan se reserva para casos severos o refractarios.
En los casos de rápida progresión o enfermedad refractaria, se puede plantear tratamiento biológico con Rituximab o Tocilizumab.
El trasplante de progenitores hematopoyéticos está dando buenos resultados en pacientes adultos.
En comparación con adultos, los pacientes con esclerodermia juvenil tienen mejores resultados y menor mortalidad(17).
Tablas y figuras
Tabla I. Criterios de clasificación del Síndrome Antifosfolípido ACR/EULAR 2023
|
Criterios de Entrada
Al menos un criterio clínico (D1-D6)
+
Un resultado de anticuerpos antifosfolípidos positivo
Anticoagulante lúpico, títulos moderados o altos de anticardiolipina o anti-β2 glicoproteína (lgG o lgM)
dentro de los tres años del criterio clínico.
|
|
Si los criterios de entrada son positivos aplique los criterios adicionales.
Criterios clínicos y de laboratorio adicionales
No tenga en cuenta un criterio clínico si existe una explicación con igual o mayor probabilidad diferente
a Síndrome antifosfolípido.
En cada dominio, solo cuente el criterio de más peso para la suma total.
|
|
Dominio clínico y criterio
|
Peso
|
Dominio clínico y criterio
|
Peso
|
|
D1. Macrovascular
(Tromboembolismo venoso (TVP))
|
D2. Macrovascular
(Trombosis arterial (TA))
|
- Con alto perfil de riesgo para TVP
|
1p
|
- Con alto perfil de riesgo de enfermedad cardiovascular
|
2p
|
- Con bajo perfil de riesgo para TVP
|
3p
|
- Con bajo perfil de riesgo de enfermedad cardiovascular
|
4p
|
|
D3. Microvascular
|
D4. Obstétrico
|
|
Sospecha de uno o más de los siguientes:
- Livedo racemosa (EF)
- Vasculopatía livedoide (EF)
- Nefropatía aPL aguda/crónica (EF, lab)
- Hemorragia pulmonar (síntomas o imagen)
|
2p
|
- ≥3 muertes pre-fetales ( < 10 semanas) y/o fetales tempranas (10-15 semanas)
|
1p
|
- Muerte fetal (16-33 semanas) en ausencia de preeclampsia con características de severidad o insuficiencia placentaria con características de severidad.
|
1p
|
|
Confirmado uno o más de los siguientes:
- Vasculopatía livedoide (AP)
- Nefropatía aPL aguda/crónica (AP)
- Hemorragia pulmonar (BAL, AP)
- Enfermedad miocárdica (imagen o AP)
- Hemorragia adrenal (imagen o AP)
|
5p
|
- Preeclampsia con características de severidad (< 34 semanas) o insuficiencia placentaria con datos de severidad, con o sin muerte fetal.
|
3p
|
- Preeclampsia con características de severidad ( < 34 semanas) + insuficiencia placentaria con datos de severidad, con o sin muerte fetal.
|
4p
|
|
D5. Válvula cardiaca
|
|
D6. Hematología
|
|
|
|
2p
|
- Trombocitopenia (menor de 20-130 x 109/L)
|
2p
|
|
|
4p
|
|
Dominio de laboratorio y peso
|
|
D7. Determinación de anticuerpos antifosfolípido por ensayo funcional basado en coagulación: ensayo anticoagulante lúpico (ALE)
|
D8. Determinación de anticuerpos antifosfolípido por ensayo de fase sólida (ELISA) anticuerpos anti-Cardiolipina (aCL) y/o anticuerpos anti-β2 glicoproteína (persistente)
|
- ALE positivo (una determinación positiva)
|
1p
|
- Positivo moderado o elevado (lgM) (aCL y/o
anti β2 gliproteína)
|
1p
|
- Positivo moderado o elevado (lgG) (aCL y/o
anti β2 gliproteína)
|
4p
|
- ALE positivo (persistente)
|
5p
|
- Positivo elevado (lgG) (aCL o anti β2 gliproteína)
|
5p
|
- Positivo elevado (lgG) (anticardiolipina y anti β2 gliproteína)
|
7p
|
|
Clasificación como Síndrome antifosfolípido para propósitos de investigación si hay al menos 3 puntos en los dominios clínicos (D1-D6) y al menos 3 puntos en los dominios de laboratorio. (D7-D8)
|
Adaptado de: Barbhaiya M, Zuily S, Naden R, Hendry A, Manneville F, Amigo MC et al. ACR/EULAR APS Classification Criteria Collaborators. The 2023 ACR/EULAR Antiphosholipid Syndrome Classification Criteria. Arthritis Rheumatol. 2023 Aug 28. doi: 10.1002 art.42624.
Tabla II. Criterios para el diagnóstico de dermatomiositis juvenil
|
CRITERIOS PARA EL DIAGNÓSTICO DE DERMATOMIOSITIS JUVENIL
|
- Debilidad simétrica de la muscular proximal.
- Biopsia muscular con evidencia de necrosis, fagocitosis, regeneración, atrofia perifascicular, variación en el tamaño de las fibras musculares, infiltrado inflamatorio perivascular.
- Elevación de enzimas musculares séricas.
- Alteraciones electromiográficas demostrando evidencia de miopatía y denervación.
- Manifestaciones cutáneas características (rash heliotropo, pápulas de Gottron).
- Biopsia confirmatoria para miopatía.
|
|
Diagnóstico:
DMJ “definida”: cambios cutáneos + 3 criterios adicionales.
DMJ “probable”: cambios cutáneos + 2 criterios adicionales.
DMJ “posible”: cambios cutáneos + 1 criterio adicional.
|
Adaptados de: Bohan y Peter.
Tabla III. Anticuerpos asociados a miopatías y fenotipo clínico asociado
|
ANTICUERPOS
|
FRECUENCIA
|
FENOTIPO CLÍNICO ASOCIADO
|
|
Anti-p155/140 (TIF-1γ)
|
18-32 %
|
Afectación cutánea severa, lipodistrofia, úlceras cutáneas, edema.
|
|
Anti-MDA5
|
7-38 %
|
Afectación pulmonar severa, elevación de RFA, escasa afectación muscular, artritis, pérdida ponderal, adenopatías.
|
|
Anti-NXP2
|
15-23 %
|
Peor pronóstico, atrofia cutánea, contracturas articulares, afectación gastrointestinal, calcinosis.
|
|
Ro-52
|
14 %
|
Manos de mecánico, artritis, afectación cutánea, más riesgo de cáncer, mayor afectación muscular, peor pronóstico (curso más crónico, más afectación pulmonar intersticial).
|
|
Anti-mi2α and β
|
4-10 %
|
Mejor pronóstico, afectación muscular severa, menos afectación orgánica, buena respuesta a tratamiento inmunosupresor.
|
|
Anti-PM/Scl
|
4-5 %
|
Enfermedades autoinmunes overlap, rash, manos de mecánico, Raynaud, enfermedad pulmonar intersticial.
|
|
Anti-SRP
|
0-1.8 %
|
Miopatía necrotizante, inicio severo, Raynaud, afectación cardiaca, úlceras cutáneas, curso crónico.
|
|
Anti-PL7
|
<5 %
|
Más afectación pulmonar y más severa, complicaciones gastrointestinales.
|
|
Anti-PL12
|
Más prevalente y severa afectación pulmonar.
|
|
Anti-Jo1
|
Afectación muscular severa.
|
|
Anti-Ku
|
<1 %
|
Enfermedades overlap, artritis, fenómeno de Raynaud, enfermedad pulmonar intersticial.
|
RFA: reactantes de fase aguda.
Adaptado(13) Sener S, Basaran O, Batu ED, Sag E, Oz S, Talim B, et al. Early-onset juvenile dermatomyositis: A tertiary referral center experience and review of the literature. Semin Arthritis Rheum. febrero de 2023;58:152133.
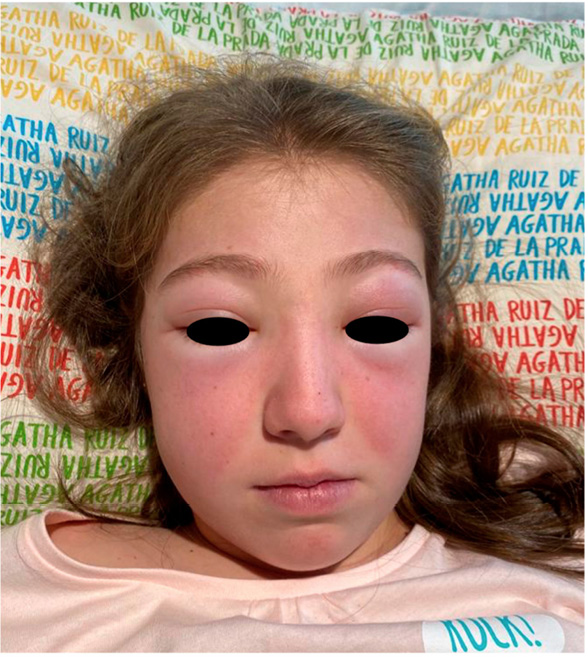
Figura 1. Lesiones cutáneas (rash malar) en el LESp
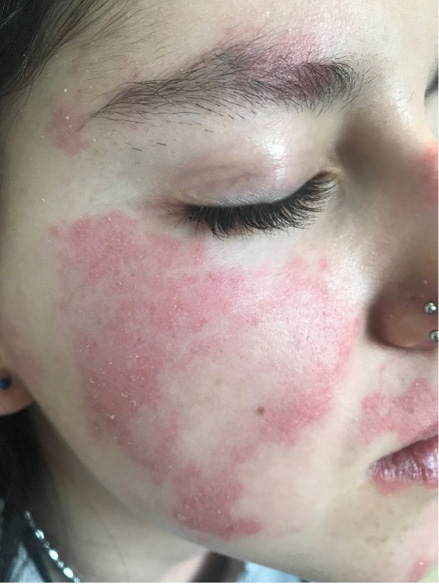
Elaboración propia.
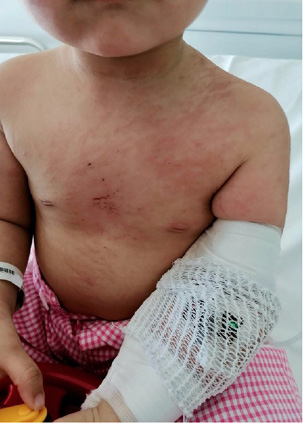
Figura 2. Afta oral en paciente con LES
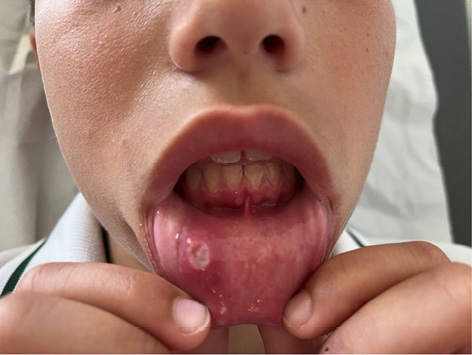
Elaboración propia.
Figura 3. Pápulas de Gottron, eritema periungueal, hipertrofia de cutículas en una adolescente con DM juvenil
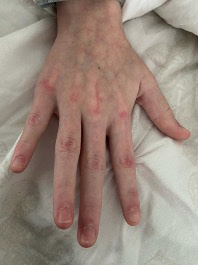
Elaboración propia.
Bibliografía
1. Smith EMD, Lythgoe H, Midgley A, Beresford MW, Hedrich CM. Juvenile-onset systemic lupus erythematosus: Update on clinical presentation, pathophysiology and treatment options. Clin Immunol. 2019;209:108274.
2. Vinuesa CG, Shen N, Ware T. Genetics of SLE: mechanistic insights from monogenic disease and disease-associated variants. Nat Rev Nephrol. 19(9):558-72.
3. Piette JC. Updating the American College of Rheumatology criteria for systemic lupus erythematosus: comment on the letter by Hochberg. Arthritis Rheum. abril de 1998;41(4):751.
4. Petri M, Orbai A, Alarcón GS, Gordon C, Merrill JT, Fortin PR, et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum. agosto de 2012;64(8):2677-86.
5. Aringer M, Costenbader K, Daikh D, Brinks R, Mosca M, Ramsey-Goldman R, et al. 2019 European League Against Rheumatism/American College of Rheumatology classification criteria for systemic lupus erythematosus. Ann Rheum Dis. septiembre de 2019;78(9):1151-9.
6. Lerkvaleekul B, Chobchai P, Rattanasiri S, Vilaiyuk S. Evaluating performance of the 2019 EULAR/ACR, 2012 SLICC, and 1997 ACR criteria for classifying adult-onset and childhood-onset systemic lupus erythematosus: A systematic review and meta-analysis. Front Med. 22 de diciembre de 2022;9:1093213.
7. Morán-Álvarez P, Andreu-Suárez Á, Caballero-Mota L, Gassiot-Riu S, Berrueco-Moreno R, Calzada-Hernández J, et al. Non-criteria manifestations in the presence of antiphospholipid antibodies in a paediatric cohort. Rheumatology.
2 de noviembre de 2022;61(11):4465-71.
8. Groot N, de Graeff N, Avcin T, Bader-Meunier B, Brogan P, Dolezalova P, et al. European evidence-based recommendations for diagnosis and treatment of childhood-onset systemic lupus erythematosus: the SHARE initiative. Ann Rheum Dis. noviembre de 2017;76(11):1788-96.
9. Miyakis S, Lockshin MD, Atsumi T, Branch DW, Brey RL, Cervera R, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost JTH. febrero de 2006;4(2):295-306.
10. Barbhaiya M, Zuily S, Naden R, Hendry A, Manneville F, Amigo M, et al. The 2023 ACR / EULAR Antiphospholipid Syndrome Classification Criteria. Arthritis Rheumatol. octubre de 2023;75(10):1687-702.
11. Islabão AG, Trindade VC, da Mota LMH, Andrade DCO, Silva CA. Managing Antiphospholipid Syndrome in Children and Adolescents: Current and Future Prospects. Pediatr Drugs. enero de 2022;24(1):13-27.
12. Gezgin Yıldırım D, Baglan E, Güngörer V, Yıldız C, Tuncez S, Bülbül M, et al. Comparison of clinical and laboratory features and treatment responses in patients with clinically amyopathic juvenile dermatomyositis and classical juvenile dermatomyositis. Int J Rheum Dis. agosto de 2023;26(8):1504-11.
13. Sener S, Basaran O, Batu ED, Sag E, Oz S, Talim B, et al. Early-onset juvenile dermatomyositis: A tertiary referral center experience and review of the literature. Semin Arthritis Rheum. febrero de 2023;58:152133.
14. Bellutti Enders F, Bader-Meunier B, Baildam E, Constantin T, Dolezalova P, Feldman BM, et al. Consensus-based recommendations for the management of juvenile dermatomyositis. Ann Rheum Dis. febrero de 2017;76(2):329-40.
15. Boyarchuk O, Kuka A, Yuryk I. Clinical and autoantibody phenotypes of juvenile dermatomyositis. Rheumatology. 31 de agosto de 2022;60(4):281-91.
16. Kobayashi I, Akioka S, Kobayashi N, Iwata N, Takezaki S, Nakaseko H, et al. Clinical practice guidance for juvenile dermatomyositis (JDM) 2018-Update. Mod Rheumatol. 3 de mayo de 2020;30(3):411-23.
17. Foeldvari I, Culpo R, Sperotto F, Anton J, Avcin T, Baildam E, et al. Consensus-based recommendations for the
management of juvenile systemic sclerosis. Rheumatology. 6 de abril de 2021;60(4):1651-8.
18. Foeldvari I, Klotsche J, Kasapcopur O, Adrovic A, Terreri MT, Sakamoto AP, et al. Differences Sustained Between Diffuse and Limited Forms of Juvenile Systemic Sclerosis in an Expanded International Cohort. Arthritis Care Res. octubre de 2022;74(10):1575-84.
19. Foeldvari I, Torok KS. Review for best practice in clinical rheumatology juvenile systemic sclerosis – Updates and practice points. Best Pract Res Clin Rheumatol. septiembre de 2021;35(3):101688.
20. Aranegui B, Jiménez-Reyes J. Morphea in Childhood: An Update. Actas Dermosifiliogr. mayo de 2018;109(4):312-22.
21. Hedrich CM, Fiebig B, Hahn G, Suttorp M, Gahr M. Presentations and treatment of childhood scleroderma: localized scleroderma, eosinophilic fasciitis, systemic sclerosis, and graft-versus-host disease. Clin Pediatr (Phila). Julio de 2011;50(7):604-14.
Bibliografía recomendada
SOBRE LUPUS ERITEMATOSO SISTÉMICO (LES):
- Smith EMD, Lythgoe H, Hedrich CM. Current views on lupus in children. Curr Opin Rheumatol. 2023 Mar 1;35(2):68-81. doi: 10.1097/BOR.0000000000000913. Epub 2022 Oct 25. PMID: 36286724.

SOBRE SÍNDROME ANTIFOSFOLIPÍDICO (SAF):
- Groot N, de Graeff N, Avcin T, Bader-Meunier B, Dolezalova P, Feldman B, et al. European evidence-based recommendations for diagnosis and treatment of paediatric antiphospholipid syndrome: the SHARE initiative. Ann Rheum Dis. 2017 Oct;76(10):1637-1641. doi: 10.1136/annrheumdis-2016-211001. Epub 2017 May 4. PMID: 28473426.
SOBRE DERMATOMIOSITIS JUVENIL (DMI):
- Enders FB, Bader-Meunier B, Baildam E, Constatin T, Dolezalova P, Feldman BM, et al. Consensus-based recommendations for the management of juvenile dermatomyositis. Ann Rheum Dis 2017; 76: 329-340.
SOBRE ESCLEROSIS SISTÉMICA JUVENIL:
- Foeldvari I, Torok KS. Review for best practice in clinical rheumatology juvenile systemic sclerosis – Updates and practice points. Best Pract Res Clin Rheumatol. Septiembre de 2021;35(3):101688.
No existen conflictos de interés en la realización de este artículo.