Pubertad retrasada
Pubertad retrasada
M.T. Muñoz Calvo, J. Pozo Román
Servicio de Endocrinología. Hospital Infantil Universitario Niño Jesús. Departamento de Pediatría de la Universidad Autónoma de Madrid. CIBER Fisiopatología y Nutrición. Instituto de Salud Carlos III. Madrid
Adolescere 2013; II (2): 38-50
Caso clínico
Adolescente de 14 años y 9 meses remitida por su pediatra de atención primaria por retraso puberal
Antecedentes familiares
Madre: menarquia 12 años, Talla 155,7 cm, G2-A0-V2.
Padre: desarrollo puberal desconoce, Talla 176,4 cm.
Abuela materna: menarquia 15 años
Antecedentes personales
Embarazo normal. Parto a las 38 semanas
PRN 2500 gr, LRN 47 cm. Sin hipoglucemias. Ictericia (fototerapia unas horas)
Período neonatal normal. Cribado metabólico neonatal normal.
Desarrollo psicomotor normal.
Exploración Física
Edad: 14 años y 9 meses.
Peso: 51 kg (p25-50). Talla: 159 cm (p50). Talla diana: 160 ± 5 cm (p25-50).
Exploración:
- Tanner 2 (T2, P1, Aa)
- TA: 100/65
Resto de la exploración normal
Estudios complementarios
√ Hemograma y Bioquímica general normal
√ Estudio hormonal basal:
|
Eje Tiroideo |
|
|
TSH (µUI/ml) (vn: 0,3-5,5) |
3,03 mcUI/ml |
|
T4 libre (ng/dl) (vn: 0,6-1,4) |
0,49 ng/dl |
|
Eje gonadal |
|
|
LH (mUI/ml) (vn: 0,2-15) |
0,02 mUI/ml |
|
FSH (mUI/ml) (vn: 2-22) |
0,28 mUI/ml |
|
Estradiol (pg/ml) (vn: 10-400) |
7,4 pg/ml |
|
Test LHRH |
|
|
LH (mUI/ml) Basal: 0,02mUI/ml |
PICO: 0,19 mUI/ml |
|
FSH (mUI/ml) Basal: 0,26 mUI/ml |
PICO 0,85 mUI/ml |
|
Eje suprarrenal |
|
|
ACTH (vn: 4,7-48) |
< 1,6 pg/ml |
|
Cortisol (vn: ) |
< 1 mcg/dl |
|
Testosterona (vn: 0,01-13) |
0,01 ng/ml |
|
DHEA-S (vn: 650-3700) |
58 ng/ml |
|
Delta-4 Androstendiona (vn: 0,7-3,6) |
0,15 ng/ml |
|
Prolactina (ng/ml) (vn: 1,6-25) |
28,4 ng/ml |
|
Eje de la GH |
|
|
IGF-1 (ng/ml) (vn: 237-996) |
14 ng/ml |
|
IGFBP-3 (µg/ml) (vn: 3,2-10) |
1,79 mcg/ml |
Cariotipo: 46XX
Estudios de imagen
Ecografía abdominal: normal
Ecografía pélvica: útero de características prepuberales, medición en su eje mayor de 3,2 cm. Ovarios de 2 cm y 2,2 cm, con características ecográficas normales.
Ecografía tiroidea: ambos lóbulos presentan un tamaño pequeño, con ecoestructura alterada generalizada de componente heterogéneo
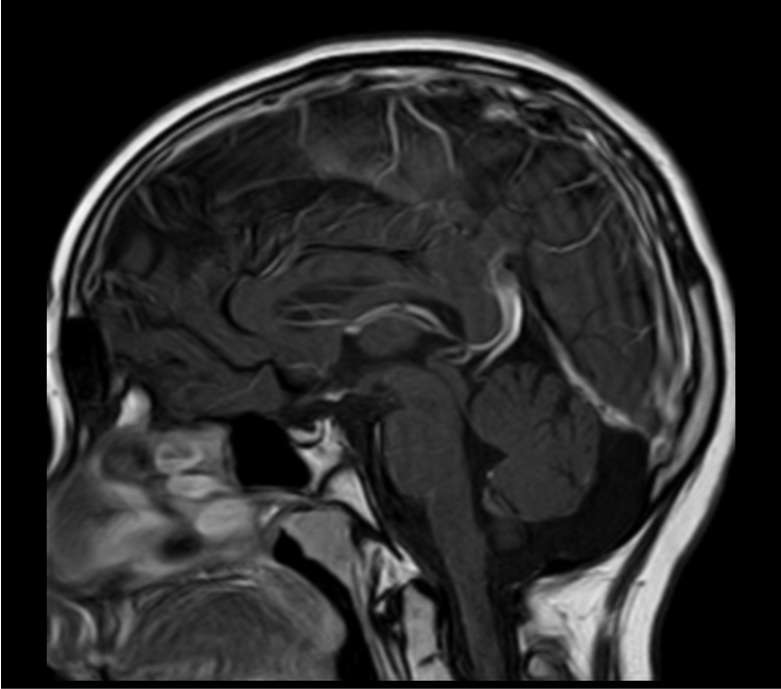
RM craneal: adenohipófisis discretamente disminuída de tamaño. No se visuliza el tallo hipofisiario. Hiperintensidad focal en secuencias T1 en localización de eminencia media, compatible con neurohipófisis ectópica (Figura 2).
Diagnóstico
Retraso puberal secundario hipogonadismo hipogonadotropo y déficit hipofisiario múltiple.
Tratamiento
• L-tiroxina: 50 µg/día
• Hidrocortisona: 5 mg a las 8 de la mañana y 5 mg a las 20 horas (8 mg/m2 sc /día)
• Estradiol transdérmico a dosis de 0,1 mcg/kg/día (Alcis® 25): ¼ de parche cada 3 días
Evolución
A los tres meses de iniciado el tratamiento con estrógenos transdérmicos, la exploración de su estadio de Tanner fue telarquia 2-3, pubarquia 1, axilarquia a.
La dosis de estrógenos se fue incrementando progresivamente, y a los 6 meses de tratamiento con una dosis actual de 12,5 mcg/día, presenta una telarquia 3, pubarquia 1, axilarquia 1. Cuando se alcance un desarrollo mamario grado IV de Tanner y un tamaño uterino por ecografía pélvica superior a 30-40 mm, con línea endometrial visible, o bien presentara sangrado vaginal recidivante, se asociará progestágenos (200 mcg/día, vo, 21 días del mes).
Concepto
No existe un consenso internacional que establezca con claridad el concepto de pubertad retrasada (PR); de hecho, el término engloba varios conceptos: PR propiamente dicha, pubertad detenida y ausencia de pubertad. Se considera la pubertad como “retrasada”, cuando no se ha iniciado el desarrollo puberal a una edad 2 SDS por encima de la edad media de su aparición en la población de referencia. A efectos prácticos, la ausencia de telarquia en las niñas a una edad de 13 años y la ausencia de incremento del volumen testicular (≥ 4 mL) en los niños a los 14 años. Se habla de pubertad “detenida” cuando la pubertad, iniciada tardíamente o no, no llega a completarse y transcurren más de 4-5 años entre su inicio y el desarrollo gonadal completo en los varones o la menarquia en las mujeres. Por último, se habla de “ausencia de pubertad”, cuando la pubertad no llega a iniciarse.
Etiopatogenia y clínica
Las causas que pueden provocar una PR son múltiples; no obstante, pueden ser fácilmente divididas en cuatro categorías (tabla I):
• Retraso puberal simple. Englobaría aquellos retrasos temporales en el inicio puberal debidos a factores constitucionales o genéticos, lo que se conoce habitualmente como: “retraso constitucional del crecimiento y de la pubertad” (RCCP).
• Retraso puberal secundario a enfermedades crónicas. Serían el resultado de trastornos funcionales en el eje hipotálamo-hipófiso-gonadal (HHG) secundarios a múltiples patologías crónicas o endocrinopatías (hipogonadismo hipogonadotropo funcional o transitorio).
• Hipogonadismos hipogonadotropos (HHipo). Serían aquellos pacientes que fracasan en su desarrollo puberal por anomalías en los mecanismos de control hipotálamo-hipofisario.
• Hipogonadismos hipergonadotropos (HHiper). Serían aquellos pacientes que fracasan en su desarrollo puberal por fallo gonadal primario.
En ambos sexos, la causa más frecuente es el simple retraso en su inicio, de etiología familiar o idiopática (RCCP), que representaría alrededor del 65 % de los casos de PR en varones y del 30 % en mujeres
Retraso constitucional del crecimiento y de la pubertad (RCCP)
Es la causa más frecuente de PR y, asociado a un componente de talla baja familiar, la causa más frecuente de talla baja en la infancia. Es más frecuente en varones (proporción de consulta de 9:1). Se considera una variante cronológica de la normalidad y puede presentarse de forma esporádica/idiopática o en un contexto familiar de maduración tardía (50-75 %). Serían niños normales con un patrón madurativo familiar más lento que la media de la población. El cuadro clínico se caracteriza por un hipocrecimiento de inicio postnatal, con un patrón de crecimiento característico y un retraso en la edad ósea (EO) y en el inicio de la pubertad de 2 a 4 años. Son niños que hasta los 12-18 meses de edad crecen normalmente, pero que, a partir de ese momento y hasta los 3-4 años, experimentan una caída en su ritmo de crecimiento que les lleva a situarse, dependiendo del componente familiar de talla, en un carril de crecimiento próximo o por debajo del percentil 3; en cualquier caso, inferior al que les correspondería para su contexto familiar. A partir de los 3-4 años, la velocidad de crecimiento (VC) se normaliza, aunque habitualmente se mantiene por debajo del percentil 50, y tienden a mantener el percentil de talla. En el periodo peripuberal, la VC disminuye de nuevo (“depresión prepuberal de la VC”), lo que los aleja nuevamente de los percentiles normales, hasta que se inicia el estirón puberal. Éste se produce tardíamente y el pico de VC suele ser menor; de forma que, es un estirón menos aparente y se ganan menos centímetros que cuando éste se produce a una edad media o temprana, compensándose así el mayor número de años de crecimiento. La talla final se alcanza también tardíamente y suele ser acorde con el contexto familiar, aunque, en alrededor del 15 % de estos pacientes, por causas desconocidas, la talla final se sitúa por debajo de lo esperable para su contexto familiar.
Retraso puberal secundario a patología crónica
Prácticamente, cualquier enfermedad crónica, si es lo suficientemente importante en gravedad y duración, repercute negativamente sobre el crecimiento y el ritmo de maduración (Tabla II). Los mecanismos fisiopatológicos que median el retraso puberal en las patologías crónicas son múltiples y varían dependiendo de la enfermedad y de la terapia empleada; no obstante, en la mayoría de ellas, existe un cierto componente de malnutrición (exceso de pérdidas, disminución de ingesta o aumento de necesidades) lo que condiciona unos mecanismos de adaptación hormonal que afectan, sobre todo, al eje de la hormona de crecimiento (GH) (resistencia parcial a la acción de la GH, retraso de crecimiento y de la maduración ósea) y al eje HHG (retraso puberal secundario a hipogonadismo hipogonadotropo funcional transitorio). Dentro de endocrinopatías que pueden asociar retraso puberal, la deficiencia aislada de GH, sobre todo parcial, puede remedar y requerir diagnóstico diferencial con el RCCP; ya que, ambas situaciones presentan importantes similitudes clínicas (hipocrecimiento y retraso en la maduración ósea), sobre todo durante el periodo peripuberal, cuando en el RCCP la VC disminuye y es frecuente observar respuestas patológicas a los test de estimulación de la secreción de GH (“deficiencia transitoria de GH”), que se supone son debidas al retraso en el incremento de esteroides sexuales (ES).
Hipogonadismos hipogonadotropos (HHipo)
Son responsables de alrededor del 10 % de los retrasos puberales. Se caracterizan por niveles muy disminuidos o ausentes de gonadotropinas circulantes, LH y FSH (tabla I).
• HHipo adquiridos. Son los más frecuentes y, en su mayoría, debidos a procesos tumorales o infiltrativos que afectan a la región hipotálamo-hipofisaria y que originan deficiencias hipofisarias múltiples bien por invasión tumoral directa, o bien como consecuencia de su extirpación quirúrgica o de la radioterapia aplicada para su tratamiento. El más frecuente de estos tumores en la infancia es el craneofaringioma, pero otros tumores, como: germinomas, gliomas o prolactinomas, pueden determinar manifestaciones clínicas similares. La dosis de radioterapia, recibida por el hipotálamo o la hipófisis, necesaria para producir un HHipo no está claramente establecida, aunque suele ser mayor de 40 Gy. Procesos infiltrativos (histiocitosis, sarcoidosis, hemocromatosis), traumatismos craneales, procesos infecciosos o inflamatorios (hipofisitis autoinmune) que afecten al área hipotálamo-hipofisaria son otras posibles causas de HHipo.
• HHipo congénitos. Su prevalencia se estima en alrededor de 1:10.000 personas y la mayoría son casos esporádicos (tabla I).
• HHipo aislados. Clásicamente se han clasificado como “HHipo congénitos con y sin alteraciones del olfato”. La asociación de HHipo congénito por deficiencia del factor liberador de gonadotropinas (GnRH) y alteración del olfato (anosmia o hipoosmia), secundaria a aplasia/hipoplasia de los bulbos olfatorios, es lo que se conoce como síndrome de Kallmann (SK). Supone alrededor de un 15 % de los HHipo y es cinco veces más frecuente en varones que en mujeres. Los pacientes con SK pueden mostrar además de los trastornos del olfato, de los que frecuentemente no son conscientes, alteraciones muy variadas, entre ellas: agenesia renal unilateral, defectos atriales septales, ceguera para los colores, hipoacusia neurosensorial, sincinesias de los dedos y lesiones de línea media. Los casos esporádicos son los más frecuentes. EL primer gen responsable del SK, el gen KAL1 (Xp22.3), responsable de las formas hereditarias ligadas al X, y codifica para una proteína, la anosmina, que facilita el crecimiento y la migración neuronal. Desde entonces, se han descrito un total de 6 genes diferentes (tabla I) asociados al síndrome, que serían responsables en conjunto de sólo un 25-35 % de los SK y con patrones hereditarios diferentes (autosómico dominante, recesivo y ligado al X).
• HHipo asociados a otras deficiencias hipofisarias. Son de causa idiopática o debidos a anomalías congénitas en el desarrollo del SNC (displasia septoóptica, holoprosencefalia…) o a mutaciones en factores de transcripción implicados en el desarrollo de las diferentes líneas células hipofisarias (tabla I).
• HHipo asociados a cuadros sindrómicos. Determinados síndromes pueden asociar cuadros clínicos de HHipo, como es el caso de los síndromes de: Noonan, Prader-Willi, CHARGE y Bardet-Biedl, entre otros.
Hipogonadismos hipergonadotropos (HHiper)
Este tipo de hipogonadismos son debidos a fallo gonadal y se caracterizan por niveles séricos elevados de gonadotropinas y disminuidos de ES. Pueden ser congénitos o adquiridos (tabla I).
• HHiper congénitos. Las dos causas más frecuente de HHiper congénito son dos cromosomopatías congénitas: el síndrome de Klinefelter y el síndrome de Turner.
El síndrome de Klinefelter o síndrome de disgenesia de los túbulos seminíferos (47, XXY y sus variantes) es la causa más frecuente de HHiper en el varón (1:500-1.000 niños nacidos vivos). La función de los túbulos seminíferos y de las células de Leydig está alterada y la espermatogénesis ausente. Dependiendo de los niveles de testosterona, la pubertad puede retrasarse o empezar a una edad normal, aunque sin una adecuada progresión. Las manifestaciones clínicas son variables, pero la talla suele ser alta y en la pubertad y edad adulta las proporciones corporales son eunucoides, el pene y los testes pequeños y la ginecomastia frecuente. Otras anomalías asociadas incluirían: retraso mental, dificultades en el lenguaje, problemas de conducta e incremento en la incidencia de determinados tumores (cáncer de mama y tumores de células germinales de localización mediastínica, retroperitoneal y pineal) y alteraciones tiroideas, entre otras.
El síndrome de Turner (45, X0 y sus variantes) es la causa más frecuente de HHiper en la mujer (1:1.500-2.500 niñas nacidas vivas). Sus manifestaciones clínicas resultan de la ausencia de genes que escapan a la inactivación del X. Estas niñas pueden presentar diferentes alteraciones y anomalías, incluyendo: hipocrecimiento, fallo gonadal, rasgos sindrómicos (pterigium colli, linfedema, tórax en coraza, hipoplasia areolar, cubitus valgo, alteraciones ungueales, acortamiento de metacarpianos, implantación baja del cabello y de las orejas, boca de pez, nevus múltiples, etc.), cardiopatía, malformaciones del sistema urinario, etc. Los síntomas más constantes son el hipocrecimiento (95 %) y el fallo gonadal (90%).
• HHiper adquiridos. Las causas son relativamente infrecuentes: torsión gonadal bilateral (testicular u ovárica), castración quirúrgica (tumores), traumatismos severos en el escroto y testículos, orquitis bilaterales (por ej. parotiditis) y, en el caso de las mujeres, galactosemia o fracaso ovárico precoz de etiología idiopática o autoinmune. El tratamiento del cáncer, debido a la quimioterapia y radioterapia, con frecuencia aplicadas conjuntamente, es una causa creciente de HHiper adquirido.
Evaluación diagnóstica
• Anamnesis detallada. Una historia familiar de PR está presente en la gran mayoría de los casos de RCCP. Un interrogatorio cuidadoso puede poner de manifiesto la presencia de síntomas sugerentes de patologías concretas (anosmia, galactorrea, hipotiroidismo, etc.) o de patologías crónicas inadvertidas, así como un exceso de ejercicio o un trastorno de la conducta alimentaria. Los antecedentes de criptorquidia, quimioterapia, radioterapia gonadal o craneal y la administración crónica o prolongada de medicamentos que puedan interferir en la función gonadal deben ser, también, recogidos. La reconstrucción de la gráfica de crecimiento y peso puede ser de utilidad. El hipocrecimiento es un hallazgo habitual en: RCCP, malnutrición, patología crónica o síndrome de Turner; mientras que, en la mayoría de las formas de HHipo la talla suele ser normal o incluso alta. Un bajo peso para la talla puede indicar trastornos nutricionales o enfermedad crónica inadvertida; mientras que: hipotiroidismo, hipercortisolismo, deficiencia de GH o determinados síndromes (Prader Willi, Turner, etc.) suelen tener un claro o moderado incremento del peso para la talla (Figura 1).
• Exploración física. Debe ser completa, analizando especialmente: signos de malnutrición o patología crónica, estigmas sindrómicos (síndrome de Turner, Klinefelter…) y signos neurológicos sugerentes de patología intracraneal (fondo de ojo, campimetría y estudio del olfato pueden ser necesarios) (Figura 1). Debe realizarse una cuidadosa valoración de los genitales y del estadio de desarrollo puberal; ya que, signos incipientes de desarrollo puberal pueden pasar inadvertidos para los pacientes o alteraciones en la secuencia normal de la pubertad pueden sugerir patología. En las niñas con desarrollo puberal normal, pero sin menarquia, deben descartarse causas anatómicas de amenorrea (himen imperforado, septum transverso vaginal o disgenesia mülleriana -síndrome de Rokitansky-), mediante una adecuada exploración ginecológica y ecográfica.
• Edad ósea (EO). El RCCP, la patología crónica, las endocrinopatías y los hipogonadismos presentan, habitualmente, un retraso de la EO de 1 a 4 años. Una talla normal-baja, con ralentización reciente de la VC y EO inferior a 11 años en una niña y a 13 años en un varón es muy sugerente de retraso puberal simple; por el contrario, la ausencia de signos puberales a una EO de más de 11 años en las niñas y de más de 13 años en los niños es muy sugerente de hipogonadismo (Figura 1).
• Otras pruebas complementarias. Su realización dependerá de la historia, exploración y EO (Figura 1). En pacientes en los que la historia clínica o la exploración física sugieran la presencia de patología crónica subyacente, debe realizarse una evaluación individual orientada a la sospecha clínica. Ésta puede incluir: hemograma y bioquímica básica, marcadores de enfermedad celíaca, TSH, T4 libre, prolactina y marcadores de deficiencia de GH (IGF-I, IGFBP-3). La sospecha de una deficiencia de GH puede obligar a realizar test de GH precedidos de la administración de ES (primación) para diferenciar una deficiencia real de GH de una deficiencia transitoria asociada a RCCP. La realización de un cariotipo estaría indicada ante la presencia de estigmas sindrómicos o en el caso de gonadotropinas elevadas, niñas con talla baja de etiología incierta o varones con testes pequeños o inadecuados para el grado de desarrollo puberal.
En la evaluación del retraso puberal, pero especialmente compleja, es la valoración del eje HHG. En la valoración del eje HHG los niveles séricos de testosterona y estradiol son de escasa utilidad en las fases iniciales de la pubertad; ya que, sus niveles séricos se sitúan, con frecuencia, por debajo del límite de detección de la mayoría de los inmunoanálisis. A partir de los 10-11 años de EO, a veces incluso antes, es frecuente observar, en los HHiper, niveles séricos elevados de LH y FSH basales o tras estímulo con GnRH. El diagnóstico de los HHipo completos también es sencillo cuando la EO supera, al menos en un año, la edad ósea en la que habitualmente la pubertad se inicia. Se observan en este caso, niveles séricos disminuidos de LH y FSH tras estímulo con GnRH.
El principal problema de diagnóstico diferencial se plantea entre el RCCP y el HHipo (sobre todo si es parcial, aislado e idiopático) cuando la EO del paciente está retrasada por debajo de las edades en que normalmente se inicia la pubertad. En estos casos, existe un considerable solapamiento entre la pobre respuesta de los pacientes con RCCP y la observada en pacientes con HHipo. En muchos casos, sólo el tiempo y la evolución espontánea de la pubertad permitirán excluir o confirmar, definitivamente, el hipogonadismo.
Tratamiento
1. Retraso constitucional del crecimiento y de la pubertad
En la gran mayoría de los casos, una clara explicación al paciente y a los padres, junto con un adecuado control y apoyo psicólogico, son suficientes. Sólo aquellos casos en los que el retraso sea más severo y existan graves repercusiones psicológicas y sociales (depresión, baja autoestima, fracaso escolar, etc.) serán susceptibles de tratamiento.
En los varones y a partir de los 12 años de EO o de los 14 de edad cronológica, puede administrarse ésteres de testosterona en preparados depot (enantato o cipionato), a dosis bajas (50-100 mg), en inyección intramuscular mensual. Por debajo de esa edad, rara vez es necesario desde el punto de vista psicológico y el riesgo de acelerar la maduración ósea y comprometer la talla definitiva es mayor. La testosterona acelera la VC, el desarrollo de los caracteres sexuales secundarios y, posiblemente también, favorece el desarrollo espontáneo de la pubertad; de hecho, en la mayoría de los pacientes el volumen testicular aumenta, lo que constituye un buen indicador de la ausencia de hipogonadismo. Se recomienda realizar ciclos de 3-6 meses, alternando con periodos similares de observación durante los cuales se vigila la progresión espontánea de la pubertad. La incidencia de RCCP en niñas es muy inferior a la de los varones y la experiencia menor. Se recomienda que el tratamiento no se inicie antes de los 13 años de edad cronológica y de los 11-12 años de EO y que se utilicen estrógenos (estrógenos conjugados o etinil-estradiol) a dosis muy bajas, al objeto de no acelerar en exceso la maduración ósea y comprometer la talla final.
En pacientes con RCCP y malas expectativas de talla adulta se ha sugerido la posibilidad de emplear otro tipo de tratamientos, como sería el caso de la GH; sin embargo, al menos con las pautas utilizadas, la GH no parece ser capaz de mejorar significativamente la talla final en estos pacientes. Otros estudios más recientes sugieren que, en varones, los inhibidores de la aromatasa de 3ª generación (letrozol y anastrozol) asociados a andrógenos podrían acelerar la VC, enlentecer la progresión de la maduración ósea y mejorar las expectativas de talla final y, todo ello, sin efectos secundarios reseñables; no obstante, los estudios al respecto son todavía muy preliminares como para poder establecer una indicación terapéutica.
2. Retraso puberal secundario a patología crónica
El tratamiento y la prevención del retraso puberal en pacientes con patologías crónicas se basa en el tratamiento óptimo y precoz de la enfermedad de base, junto con una adecuada nutrición (aporte suficiente de macro y micronutrientes). Las pautas para inducir y mantener el desarrollo puberal no difieren, en general, de las empleadas en el RCCP o en el hipogonadismo.
3. Hipogonadismos
Una propuesta aceptable sería inducir el desarrollo puberal alrededor de los 11 años de EO en las niñas y de los 12 años en los varones e incrementar lentamente los niveles séricos de ES para conseguir un desarrollo puberal completo en un periodo de 3-4 años. Cuando la talla final está comprometida (deficiencia de GH, Turner), puede ser necesario retrasar deliberadamente la inducción de la pubertad, al objeto de intentar mejorar las expectativas de talla adulta.
En varones, la forma más sencilla de inducir el desarrollo puberal es la administración de preparados depot de testosterona de acción prolongada (enantato o cipionato) por vía intramuscular. La dosis inicial de 25-50 mg c/4 semanas, se incrementará en 50 mg, c/ 6-12 meses, para, a lo largo de un periodo de 3-4 años, alcanzar la dosis de sustitución de un adulto, que oscilaría entre 200-250 mg cada 10-14 días. Un inconveniente de esta terapia es que el volumen testicular no aumenta. En los niños con HHipo en los que se desee incrementar el volumen testicular, la terapia intramuscular o subcutánea con gonadotropinas o la administración pulsátil, mediante bomba, de bolos de GnRH, por vía intravenosa o subcutánea, puede ser una alternativa, aunque sus resultados son variables y, frecuentemente poco satisfactorios. Una vez completado el desarrollo puberal, la terapia de mantenimiento en los varones se realiza, habitualmente, con testosterona, habitualmente por vía intramuscular (200-250 mg de enantato de testosterona cada 10-14 días o 1000 mg de undecanoato de testosterona c/2-3 meses) o transdérmica (parches de testosterona, cremas).
En niñas, la inducción de la pubertad se realizará con parches de estrógenos de estradiol o con estrógenos orales. Los parches transdérmicos permiten su fragmentación y la administración de dosis bajas y progresivamente crecientes de estradiol. Una vez alcanzado el desarrollo mamario y uterino deseados (telarquia 4 y tamaño uterino por ecografía pélvica de > 34-40 mm, con línea endometrial visible) o bien si existe sangrado vaginal recidivante, se asociará tratamiento con progestágenos. Los estrógenos de administración transdérmica presentan mayor biodisponibilidad que por vía oral, mejor tolerancia gastrointestinal y menor toxicidad hepato-biliar al evitar el paso inicial por el hígado, minimizando así la influencia de los estrógenos sobre el metabolismo hepático.
Los estrógenos orales se inician con dosis muy bajas; ya que, los estrógenos son un potente inductor de la fusión epifisaria. Los regímenes más habitualmente empleados incluyen la administración oral de: estrógenos conjugados equinos (0,15 mg/día o 0,3 mg a días alternos), etinil-estradiol (2,5-5 µg/día) o 17 ß-estradiol (5 µg/kg/día). Esta dosis inicial se incrementará lentamente, cada 6-12 meses, durante un periodo no inferior a 2-3 años, hasta alcanzar la dosis diaria de sustitución estrogénica de una mujer adulta, que correspondería a 0,6-1,2 mg de estrógenos conjugados equinos, 10-20 µg de etinil-estradiol o 1-2 mg/día de 17 ß-estradiol. Si se produce sangrado menstrual, pequeños manchados o cuando se lleven 6 meses de tratamiento con una dosis de estrógeno oral equivalente a 0,6 mg de estrógenos conjugados equinos, debe añadirse un progestágeno cíclico (12-14 días de cada mes) para la protección uterina y establecer ciclos menstruales regulares mensuales.
Bibliografía
1. Harrington J, Palmert MR. Clinical review: Distinguishing constitutional delay of growth and puberty from isolated hypogonadotropic hypogonadism: critical appraisal of available diagnostic tests. J Clin Endocrinol Metab. 2012;97:3056-67.
2. Fenichel P. Delayed puberty. Endocr Dev. 2012;22:138-59.
3. Palmert MR, Dunkel L. Clinical practice. Delayed puberty. N Engl J Med. 2012 2;366(5):443-53.
4. Pozo J, Argente J. Ascertainment and treatment of delayed puberty. Horm Res 2003; 60 (suppl 3): 35-48.
5. Viswanathan V, Eugster EA. Etiology and treatment of hypogonadism in adolescents. Pediatr Clin North Am. 2011;58:1181-200.
6. Wehkalampi K, Widén E, Laine T, Palotie A, Dunkel L. Patterns of inheritance of constitutional delay of growth and puberty in families of adolescent girls and boys referred to specialist pediatric care. J Clin Endocrinol Metab. 2008;93:723-8
7. Butenandt O, Kunze D. Growth velocity in constitutional delay of growth and development. J Pediatr Endocrinol Metab. 2010;23:19-25.
8. Segal TY, Mehta A, Anazodo A, Hindmarsh PC, Dattani MT. Role of gonadotropin-releasing hormone and human chorionic gonadotropin stimulation tests in differentiating patients with hypogonadotropic hypogonadism from those with constitutional delay of growth and puberty. J Clin Endocrinol Metab. 2009; 94:780-5.
9. Wehkalampi K, Päkkilä K, Laine T, Dunkel L. Adult height in girls with delayed pubertal growth. Horm Res Paediatr. 2011;76:130-5.
10. Salehpour S, Alipour P, Razzaghy-Azar M, Ardeshirpour L, Shamshiri A, Monfared MF, Gharib A. A double-blind, placebo-controlled comparison of letrozole to oxandrolone effects upon growth and puberty of children with constitutional delay of puberty and idiopathic short stature. Horm Res Paediatr. 2010;74:428-35.
11. Barrio R, de Luis D, Alonso M, Lamas A, Moreno JC. Induction of puberty with human chorionic gonadotropin and follicle-stimulating hormone in adolescent males with hypogonadotropic hypogonadism. Fertil Steril. 1999;71:244-8.
12. Aydogdu A, Bolu E, Sonmez A, Tasci I, Haymana C, Acar R, Meric C, Taslipinar A, Ozgurtas T, Azal O. Effects of Three Different Medications on Metabolic Parameters and Testicular Volume in Patients with Hypogonadotropic Hypogonadism: 3-Year Experience. Clin Endocrinol (Oxf). 2012: 27. doi: 10.1111/cen.12135.
Tablas y figuras
Tabla I. Etiopatogenia de la pubertad retrasada
|
Retraso puberal simple (RCCP) |
|
|
|
|
Retraso puberal 2rio a patologías crónicas |
|
|
Hipogonadismos hipogonadotropos (HH): |
Hipogonadismos hipergonadotropos: |
|
|
|
T1: testosterona. SIPA: síndrome de insensibilidad periférica a los andrógenos. FSHR: gen del receptor de FSH. LHR: gen del receptor de LH. S: síndrome. SNC: sistema nervioso central |
|
Tabla II. Principales patologías crónicas responsables de retraso puberal
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Figura 1. Orientación diagnóstica de la pubertad retrasada (PR)
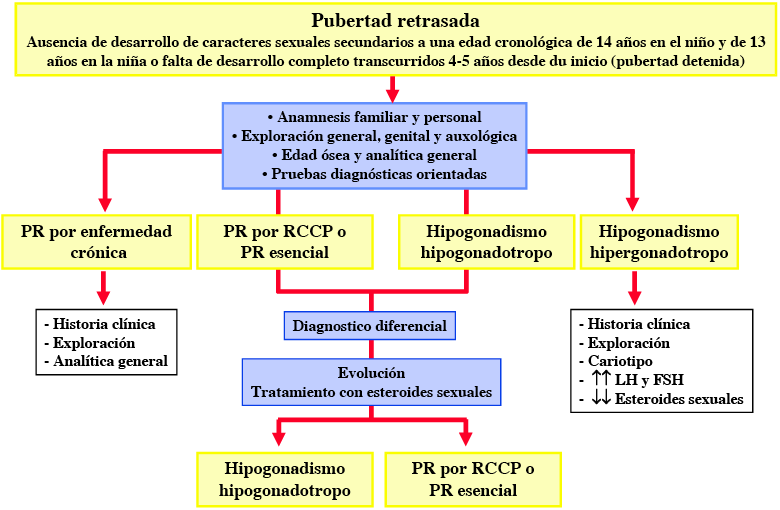
Abreviaturas: PR: pubertad retrasada; RCCP: retraso constitucional del crecimiento y de la pubertad; E.: Esteroides.
Figura 2. Resonancia craneal