Síndrome Klipper-Feil en la adolescencia: cuándo una radiografía te cambia la vida
Síndrome Klipper-Feil en la adolescencia: cuándo una radiografía te cambia la vida
L. Liquete Arauzo(1), E. Pérez Ruiz(2), A. de la Pinta Zazo(3), R. Pérez Lázaro(4).
(1)Pediatra. Centro de Salud de Villamuriel de Cerrato. Palencia, (2)Psiquiatra. Hospital Clínico Universitario de Valladolid, (3)Residente de Medicina de Familia y Comunitaria. Hospital Río Carrión de Palencia, (4)Residente de cuarto año de Radiología. Hospital Río Carrión de Palencia.
Fecha de recepción: 14-12-2021
Fecha de publicación: 28-02-2022
Adolescere 2022; X (1): 84-90
Resumen
|
Mujer de 12 años adoptada de origen chino que consulta por una cervicalgia de larga evolución. Antecedentes: pulgar bífido derecho intervenido. Múltiples ingresos e interconsultas durante la infancia. A la exploración: movilidad cervical reducida, especialmente la rotación. En las radiografías cervicales se constata una fusión completa de C6 y C7. El reto del diagnóstico de esta paciente solo acaba de empezar…. Palabras clave: Cervicalgia; Síndrome Klippel-Feil; Malformación congénita; Fusión cervical; Origen aberrante de la arteria pulmonar |
Abstract
|
Adopted 12-year-old female of Chinese origin consulting for long-standing chronic cervical pain. Personal history: Malformation in the right thumb already operated on. Several admissions and multiple consultations throughout her childhood. The physical examination of the patient shows a reduction in the mobility of the cervical spine with greater involvement of the rotational movement of the cervical spine. The cervical x-rays show a complete fusion of the cervical vertebrae C6 and C7. The challenge of diagnosing this patient has only just begun… Key words: Cervicalgia; Klippel-Feil syndrome; Congenital malformation; Cervical fusion; Pulmonary artery sling. |
Introducción
El síndrome de Klippel-Feil es una anomalía congénita caracterizada por la fusión de, al menos, dos vértebras cervicales
El síndrome de Klippel-Feil (SKF) es una anomalía congénita caracterizada por la fusión de, al menos, dos vértebras cervicales originada por una anomalía en la segmentación de las somitas cervicales entre la 3ª y la 8ª semana de gestación. La no segmentación congénita de las somitas cervicales en el SKF conduce a una formación deficiente de las vértebras y de los espacios vertebrales implicados, pudiendo generar un compromiso del canal medular. De hecho, en el SKF toda la columna cervical ve alterada su dinámica de movimiento, así las vertebras fusionadas de un tramo se comportan como un bloque rígido que transfiere las fuerzas como brazos largos de palanca a los segmentos móviles adyacentes de la columna ocasionándoles una hipermovilidad. Esta hipermovilidad puede conllevar, en determinados casos de SKF, a una inestabilidad vertebral que facilitarían la compresión de la médula espinal, las radiculopatías e incluso la muerte(1-4).
Clasificación
El SKF fue descrito por primera vez en 1912 y desde entonces se han propuesto numerosas clasificaciones, entre ellas, por su utilidad al diagnóstico, destacamos las propuestas por Nagib(1-3). y Samartzis(4).
Nagib clasifica el SKF en tres tipos en base al riesgo de asociar o/y provocar lesiones neurológicas:
Tipo I – Dos conjuntos de vértebras en bloque con espacios intermedios abiertos que pueden subluxarse de forma aguda o degenerativa. Tipo II – Anomalías craneocervicales con occipitalización del eje (asociadas a de Arnold-Chiari y siringomielia) y Tipo III – Fusión de uno o más niveles con estenosis espinal asociada (1-3). Samartzis hace una clasificación radiográfica de la fusión espinal que nos permite conocer el riesgo de síntomas radiculopáticos o mielopáticos (que es mayor entre los tipos III y II). Tipo I – Cervical única. Tipo II – Múltiples segmentos no contiguos; Tipo III – Múltiples fusiones cervicales contiguas(4).
Con el uso de estas escalas y un accesible estudio dinámico de la columna cervical podemos etiquetar un SKF como de alto/medio o bajo riesgo de lesión neurológica y actuar en consecuencia.
Clínica
El SKF se asociaría a la triada de cuello corto, rigidez cervical y a una línea de implantación baja del cabello posterior
Desde el punto de vista teórico todo SKF se asociaría a la triada de cuello corto, rigidez cervical y a una línea de implantación baja del cabello posterior. Sin embargo, menos del 50% de pacientes con SKF presentan estas tres manifestaciones.
De hecho, en la primera década de la vida, más del 50% de los niños con SKF están asintomáticos. Se ha postulado que cuanto mayor es el número de vertebras fusionadas distales a C3, mayor será la intensidad y la precocidad de aparición de los síntomas axiales (dolor de cuello, dolor de cabeza, rigidez de nuca) en el SKF. Los síntomas axiales suelen aparecer en la adolescencia temprana y empeoran con los años. Por el contrario, los síntomas radiculopáticos o mielopáticos, propios de los SKF tipo II y III de Samartzis, suelen aparecer en la adolescencia tardía(1-4).
Etiología
Las causas del SKF son desconocidas, múltiples (mutaciones genéticas, regresión precoz de las arterias vertebrales primitivas, interrupción en el desarrollo de la arteria subclavia, disturbio en el tubo neural primitivo…) y de interacción variable (el SKF fue inicialmente catalogado como un síndrome, después como una secuencia y posteriormente como una anomalía(1-6). El SKF puede presentarse de forma aislada y se postula que las mutaciones en los genes GDF6 y GDF 3, ambos implicados en el desarrollo óseo, generarían un SKF con herencia autosómica dominante; mientras que las mutaciones en el gen MEOX1, que regula la separación de las somitas, daría lugar a un SKF con herencia autosómica recesiva. El estudio genético es de utilidad en el caso de que existan varios miembros de una misma familia afectos o cuando el paciente desea evitar una posible trasmisión a su descendencia(1-3). No obstante, cuando el SKF forma parte de un cuadro clínico polimalformativo, la causa etiológica del SKF es la que genera el propio síndrome(1-6).
El SKF se diagnostica con una radiografía simple de la columna cervical. Sin embargo, ante todo SKF, es necesario realizar un estudio dinámico de la columna cervical para valorar el funcionamiento dinámico de la columna y su estabilidad. Si existe sospecha de lesión neurológica acompañante o alto riesgo de lesión medular debe de realizarse una RMN cervical(1-3).
El SKF se diagnostica con una radiografía simple de la columna cervical
Con todo, la trascendencia del diagnóstico de un SKF en un paciente, también, radica en conocer su robusta asociación con otras malformaciones congénitas. Así al diagnóstico del SKF, y su clasificación adecuada, deben seguirle un concienzudo interrogatorio, un minucioso examen clínico y la solicitud de las pruebas de imagen e interconsultas que se requieran para descartar otras malformaciones acompañantes. Las más frecuentemente asociadas son escoliosis (60%), anomalías renales (35%), deformidad de Sprengel (30%), hipoacusia (30%), sincinesias (20%) y anomalías cardiacas (15%)(1-4).
Pronóstico y tratamiento: el tratamiento del SKF está condicionado por los trastornos derivados de la fusión vertebral y por la existencia de otras malformaciones. La cirugía del SKF es necesaria si la columna cervical es potencialmente inestable y existe riesgo de lesión medular (I y II de Nagib)(1-4).
El tratamiento y pronóstico del SKF está condicionado por los trastornos derivados de la fusión vertebral y por la existencia de otras malformaciones
Caso clínico
Anamnesis
Mujer de 12 años que acude a consulta de Pediatría de Atención Primaria por dolor y rigidez cervical de meses de evolución. Antecedentes personales: adopción de origen chino. Se desconocen antecedentes. A su llegada a España con 10 meses, presentaba una somatometria normal y una malformación en el pulgar derecho que le impedía hacer pinza. Se solicitaron varias pruebas, entre ellas una radiografía de mano derecha que confirmó la existencia de una doble falange en el pulgar derecho (Wassel III) que se derivó a Traumatología. La familia consulta en varias ocasiones porque la menor se saciaba con cantidades muy pequeñas de comida sin presentar atragantamientos. Entre los 2 y 5 años presentó apneas nocturnas de repetición sin hipertrofia adenoidea asociada. Episodios de broncoespasmo de repetición con buena respuesta a broncodilatadores. Múltiples alergias ambientales.
Exploración física
Somatometría normal, saturación de oxigeno del 99% y auscultación cardiopulmonar normal. A nivel cervical, presentaba un cuello corto con movilidad limitada e hiperlordosis. Imposibilidad para realizar la maniobra de Adams.
Pruebas complementarias
- Radiografías cervicales -estáticas y dinámicas-: fusión completa de los cuerpos vertebrales, arcos posteriores y apófisis espinosas de C6 y C7. Disminución de altura del disco intervertebral C5-C6 y C7-T1 con fusión de sus arcos posteriores. Variaciones en forma y tamaño de los agujeros de conjunción que coinciden con las salidas de las raíces de C5 hasta T1.
Juicio clínico
Klipper Feil tipo III de Nagib/ Samartzi I (Figura 1A). Radiografía posteroanterior de columna vertebral. Disminución de la vascularización pulmonar izquierda con hilio pulmonar izquierdo hipoplásico (Figura 1B).
Evolución clínica
Ante el diagnóstico de SKF se realiza una exploración neurológica completa que resultó normal. Se comunica a la paciente la contraindicación de realizar actividades que exijan la hiperflexión cervical y se amplía el estudio diagnóstico para descartar otras malformaciones. Con este fin, se realizan varias interconsultas (IC) así como una ecografía abdomino-genitourinaria. Los resultados se detallan a continuación:
- Interconsulta a Cardiología Pediátrica: ECO-2D-DOPPLER cardíaco: cavidades cardíacas normales. Se visualiza tronco pulmonar (TPA) y arteria pulmonar derecha (APD). Jet de ductus persistente mínimo en TPA. No se visualiza la arteria pulmonar izquierda (API) por lo que se solicita resonancia magnética (RMN) torácica.
- RMN torácica “Sling” de la API con origen en la cara posterosuperior de la APD abrazando el margen inferolateral derecho de la tráquea en la zona previa a su bifurcación carinal. La API anómala discurre posteriormente a la tráquea y por delante del esófago, observándose en dicho punto un estrechamiento significativo de la luz arterial de la API que alcanza hasta 2 mm de diámetro. Después, la API transcurre por delante de la aorta torácica descendente y posterosuperiormente al bronquio principal izquierdo (BPI) (Fig. 2b).
Significativo retraso en la opacificación del árbol arterial pulmonar (hipovascularización) del hemitórax izquierdo. Dilatación del TPA que alcanza los 21,2 mm. Quiste de 16,3 x 10,6 x 18,6 mm en lóbulo tiroideo.
- IC Traumatología: No constan anotaciones ni pruebas complementarias en su historia clínica al respecto.
- IC Otorrinolaringología: se descarta una hipoacusia asociada.
- IC Ginecología y ecografía abdominal: se descartan malformaciones genitourinarias asociadas.
Ante los hallazgos descritos en la RMN se derivó a la paciente a la unidad de Cirugía Cardiovascular infantil de otro hospital. Tras analizar el caso, el cirujano cardiovascular infantil concluye la necesidad de realizar a la paciente una fibrobroncoscopia con el objetivo de conocer la existencia de malformaciones traqueobronquiales, asociadas al sling de API, que pudieran requerir cirugía. La familia de la paciente no está convencida de proseguir con las pruebas y decide, en contra de criterio clínico, aplazar la realización de la fibrobroncoscopia y, en consecuencia no se tramitan las consultas posteriores de seguimiento. La realización de la fibrobroncoscopia se retrasa hasta 5 años después de su petición inicial. Ya con 17 años la paciente, en consenso con la familia, solicita proseguir con los estudios de despistaje de malformaciones asociadas a su SKF. La fibrobroncoscopia mostró la ausencia de malformaciones traqueobronquiales asociadas, que de haber existido, hubieran requerido cirugía.
- IC endocrino infantil: realizaron un seguimiento continuado hasta su transición a endocrinología de adultos del bocio tiroideo multinodular con autoinmunidad tiroidea positiva. Tras realizarle varias PAAF, se decide con 16 años practicarle una tiroidectomía total. Actualmente recibe tratamiento sustitutivo de su hipotiroidismo postquirúrgico con buen control.
Diagnóstico
Cuadro polimalformativo congénito formado por:
- Klipper Feil tipo III de Nagib/ Samartzi I (Fig. 1A).
- Origen aberrante de la Arteria Pulmonar Izquierda (sling de API) (Fig. 2B).
- La polidactilia preaxial unilateral (pulgar bífido – Wassell III).
Diagnóstico diferencial y evolución clínica de la paciente
El SKF es una anomalía congénita cuya manifestación radiológica es la fusión de 2 o más vertebras cervicales. En esta paciente, cuando se diagnosticó un SKF se estimó se trataba de una lesión con bajo riesgo de lesiones neurológicas asociadas(1-4) por lo que se priorizó el despistaje de otras malformaciones congénitas asociadas. En esta paciente hallamos, además, una malformación vascular (sling de API) y otra de extremidades (polidactilia preaxial unilateral).
El pronóstico del sling de API depende del compromiso traqueal y de la existencia de malformaciones traqueobronquiales o/y cardíacas asociadas
El sling de API es una forma infrecuente de anillo vascular en la que la API tiene un origen aberrante en nuestra paciente en la APD para alcanzar posteriormente el hilio pulmonar izquierdo. El pronóstico del sling de API depende del compromiso traqueal y de la existencia de malformaciones traqueobronquiales o/y cardíacas asociadas. En el caso de nuestra paciente, la API aberrante presenta la peculiaridad de no comprimir el bronquio principal derecho ni la tráquea en su trayecto inicial lo que podría justificar, a diferencia de otros sling de API, su presentación asintomática. El sling de API es una anomalía en la embriogénesis ocasionada por una regresión precoz del 6º arco aórtico proximal izquierdo que conllevaría a una vascularización anómala del pulmón izquierdo a partir de vasos anómalos procedentes del 6º arco aórtico proximal derecho(5-6).
La polidactilia preaxial unilateral que presentaba esta paciente fue diagnosticada a su llegada a España. La polidactilia preaxial es una anomalía congénita, que puede presentarse aislada o asociada a otras malformaciones, ocasionada por una noxa teratogénica que acontecería simultáneamente a las otras malformaciones que presentaba nuestra paciente(6-8).
Con todo, el reto diagnóstico de este caso clínico de SKF no concluye aquí, sino que con los resultados de las exploraciones solicitadas se trató de clasificar a la paciente en alguno de los cuadros clínicos polimalformativos que se asocian al SKF.
En este caso, se estimó inicialmente que podría tratarse de un VACTERL, acrónimo que agrupa una serie de malformaciones congénitas asociadas a nivel vertebral, anal, cardíaco (15%), traqueoesofágico, renal (35%) y de extremidades. Para que un paciente sea diagnosticado de VACTERL debe padecer al menos 3 de las mencionadas malformaciones que componen su acrónimo. Así, esta paciente tenía anomalías vertebrales, cardíacas y de extremidades. No obstante, cuando revisamos las malformaciones cardíacas y de extremidades propias del VACTERL, hallamos que eran diferentes de las que la paciente padecía. El VACTERL se asocia a malformaciones cardíacas menores y que afectan especialmente al septo interventricular pero no al sling de API. Asimismo, las malformaciones de extremidades propias del VACTERL suelen ser anomalías radiales, incluyendo aplasia/hipoplasia del pulgar, pero no una polidactilia.
Así, esta paciente carecía de los criterios básicos para ser considerada un VACTERL y requería plantearse otras opciones diagnósticas como el síndrome de Baller-Gerold (craneosinostosis e hipoplasia del radio), el CHARGE (atresia de coanas, enfermedades cardíacas cianosantes…), el 22q11.2 (defectos cardíacos, palatinos, dimorfismo facial e inmunodeficiencia), el síndrome óculo-dígito-esofágico-duodenal u ODED (microcefalia, baja estatura y anomalías digitales), el síndrome de Fryns (dismorfia facial, hernia diafragmática congénita, hipoplasia pulmonar y de las extremidades distales), la MURCS (aplasia del conducto mulleriano, displasia renal y anomalías de los somitas cervicales), el síndrome oculoauriculovertebral (baja estatura, anomalías vertebrales, pérdida auditiva, dismorfia facial y anomalías radiales, como la polidactilia preaxial)…(1-8).
Tras una exhaustiva revisión de los cuadros malformativos asociados al SKF, se concluye que la paciente no cumplía criterios diagnósticos suficientes para ser etiquetada en ninguno de los síndromes polimalformativos conocidos(1-8).
Respecto a la indicación de realizar estudio genético en esta paciente queremos destacar que la presentación fenotípica de SKF, sling de API y polidactilia preaxial unilateral no se ha descrito, a día de hoy, como atribuible a una determinada mutación genética. Por otra parte la posibilidad de que un paciente presente simultáneamente tres secuencias malformativas y que no exista un nexo común entre ellas parece poco probable, máxime cuando coinciden las tres en el mismo momento de la embriogénesis. En base a los argumentos expuestos, creemos que este caso clínico podría constituir un síndrome polimalformativo con entidad propia (SKF, sling API y polidactilia preaxial unilateral) no descrito previamente en la literatura médica.
Tratamiento
El pulgar bífido derecho (Wassel II) fue remitido e intervenido precozmente por el servicio de Traumatología del hospital de referencia. Sin embargo, el resultado funcional del pulgar derecho no fue satisfactorio a las expectativas. Queremos subrayar, que pese a usar una técnica correcta, un pulgar reconstruido nunca tendrá el aspecto, volumen ni movilidad de un pulgar normal. El resto de malformaciones no han requerido tratamiento hasta el momento actual.
Evolución clínica
Tras su llegada a España, la paciente fue intervenida por la malformación del pulgar derecho. A lo largo de su infancia, la paciente precisó varios ingresos hospitalarios por diversos motivos (crisis de asma, apneas, síncopes de repetición…), fue sometida a variadas pruebas diagnósticas (estudio del sueño, pruebas de alergia…) e incluso fue intervenida quirúrgicamente en otras 2 ocasiones, lo que estresó mucho a los progenitores adoptivos de la menor. Cuando, finalmente, con 12 años es diagnosticada de un SKF y de un sling de API, la reacción de la familia adoptiva y de la paciente fue la negación del problema.
Paralelamente a estos hallazgos, se detecta a la paciente un nódulo tiroideo que progresa rápidamente. En el plazo de apenas 12 meses, el nódulo se convierte en un bocio multinodular que comprime estructuras adyacentes y que, finalmente, requiere una tiroidectomía total a los 16 años de vida.
La negación del problema por parte de la familia, unida al cambio de profesional de referencia (al cumplir 14 años pasó de Pediatría a Medicina de Familia) y a la imperante necesidad de controlar su bocio eclipsó, a nuestro juicio, la necesidad de un seguimiento multidisplinar adecuado así como de un estudio de imagen de alta resolución que evalúe si existe compromiso de estructuras vasculonerviosas a nivel cervical con el objetivo de evitar lesiones medulares y nerviosas en un futuro. En el momento actual, la paciente es mayor de edad y se han reanudado las pruebas solicitadas y manejo multidisplinar.
Discusión y conclusiones
El diagnóstico de un SKF es todo un desafío para el pediatra o médico que lo diagnostica. En primer lugar, porque es una malformación congénita que, con frecuencia, se diagnostica fuera del periodo neonatal, y que se asocia a otras malformaciones congénitas, siendo el punto de partida para múltiples valoraciones interdisciplinares. En segundo lugar, este caso nos muestra las dificultades en el seguimiento clínico y en la prevención de complicaciones que acontecen tras el diagnóstico demorado o causal de malformaciones congénitas de escasa manifestación clínica, y reivindica la necesidad de un médico de referencia estable que realice la coordinación entre los múltiples especialistas hospitalarios que atienden al paciente. Muestra, además, la necesidad imperante de crear guías o algoritmos decisorios personalizados que permitan al clínico prevenir y tratar las plausibles complicaciones derivadas de un cuadro polimalformativo aún no catalogado; así como de conocer la posibilidad de recurrir a la figura del menor maduro ante la negativa de la familia a asumir el diagnóstico. En último lugar destacamos que este caso clínico constituye un síndrome polimalformativo con entidad propia y no publicado previamente en la literatura médica, y, que incluye al mismo tiempo, una secuencia malformativa infrecuente (sling de API).
Conflicto de intereses:
Los autores declaran no tener ningún conflicto de interés.
Tablas y figuras
Figura 1a. Radiografía cervical lateral 1b. Radiografía PA tórax
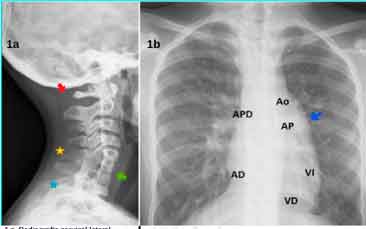
1a) Radiografía posteroanterior de columna vertebral. Estrella amarilla: fusión parcial C5-C6.
- Estrella azul: fusión parcial C7-T1.
- Flecha verde: fusión completa C6-C7.
- Flecha roja: integridad unión cráneo-cervical.
1b) Disminución de la vascularización pulmonar izquierda con hilio pulmonar izquierdo hipoplásico.
Figura 2. Comparativa entre RMN normal (2a) y
la de la paciente (2b)
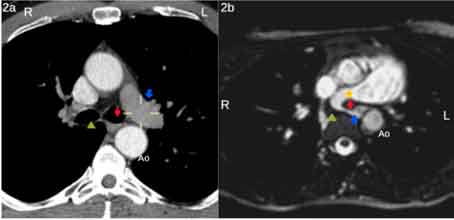
2a) RMN normal:
- Flecha azul: arteria pulmonar izquierda
2b) RMN paciente:
- Estrella amarilla: arteria pulmonar derecha
- Flecha azul: origen aberrante de la arteria pulmonar izquierda (sling)
- Rombo rojo: bronquio principal izquierdo
- Cabeza flecha verde: esófago
Bibliografía
1. Menger RP, Rayi A, Notarianni C. Klippel Feil Syndrome. 2021. StatPearls [Internet]. [Consultado 10 Mar 2022]. PMID: 29630209. Disponible en: https://pubmed.ncbi.nlm.nih.gov/29630209/.
2. Hachem L, Mathieu F, Lamberti-Pasculli M, Hanak B, Zeller R, Kulkarni AV et al. Klippel Feil Syndrome. SPINE. 2020; 45(11): 718-26.
3. Vaidyanathan S, Hughes PL, Soni BM, Singh G, Sett P. Klippel-Feil syndrome – the risk of cervical spinal cord injury: a case report. BMC Fam Pract. 2002. 2293- 6.
4. Samartzis DD, Herman J, Lubicky JP, Shen FH. Classification of congenitally fused cervical patterns in Klippel-Feil patients: epidemiology and role in the development of cervical spine-related symptoms. Spine. 2006; 31(21):798-804.
5. Bueno J, Flors L, Mejía M. Anomalías congénitas de las arterias pulmonares: espectro de hallazgos en tomografía computarizada. Radiología. 2017; 59(3):209-217.
6. Cruz M, Bosch J. Atlas de síndromes pediátricos. Espaxs editorial. Barcelona. 1998.
7. Da-Silva EO. Preaxial polydactyly and other defects associated with Klippel-Feil anomaly. Hum Hered. 1993 Nov-Dec;43(6):371-4. doi: 10.1159/000154162. PMID: 8288268.
8. Urdaneta E, Rojas G, Urdaneta A, Maldonado M, Brito M. Síndrome de Klippel-Feil con fístula traqueoesofágica, pulgar bífido y angiolipoma cerebral. Arch. argent. pediatr. [Internet]. 2016; 114(6): 398-402.
Bibliografía recomendada
- Lewis TR. Klippel-Feil Syndrome. Orthopedic Surgery Medscape. [Internet]. 2019. [Consultado el 10 de marzo de 2022]. disponible en: http://emedicine.medscape.com/article/1264848-overview.
- Doménech C, Doménech P. Dolor lumbar, dorsal y espondilolisis en el adolescente. Adolescere 2021; IX (3): 37-47.

