Manifestaciones respiratorias en la fibrosis quística
Manifestaciones respiratorias en la fibrosis quística
M.M. Mendoza Chávez.
Servicio de neumología pediátrica HM Hospitales. Madrid. Servicio de neumología pediátrica Hospital Rey Juan Carlos.
Fecha de recepción: 31 de agosto 2018
Fecha de publicación: 15 de octubre 2018
Adolescere 2018; VI (3): 52.e1-52.e8
Resumen
|
La fibrosis quística es la enfermedad autosómica recesiva más común en la población caucásica y es causada por mutaciones en un gen en el cromosoma 7 que codifica la proteína reguladora de la conductancia transmembrana de la fibrosis quística (CFTR). La alteración en la proteína CFTR resulta en secreciones espesas y viscosas en los bronquios, las vías biliares, el páncreas, los intestinos y el sistema reproductivo que afectan su función normal. Los signos y síntomas de la enfermedad pulmonar son muy diferentes y van desde lactantes asintomáticos detectados por cribado neonatal a niños mayores y adolescentes con tos productiva crónica, colonización crónica por patógenos como P. aeruginosa, presencia de bronquiectasias y alteración significativa de la función pulmonar. El tratamiento se basa en lograr una nutrición adecuada, reducir la obstrucción bronquial y promover el aclaramiento mucociliar a través de la fisioterapia respiratoria, el ejercicio y el tratamiento precoz de las infecciones respiratorias. Recientemente, se están incorporando al tratamiento terapias específicas que actúan mejorando la producción, el procesamiento intracelular y/o la función de la proteína CFTR defectuosa, lo que revolucionará el pronóstico de esta enfermedad.. Palabras clave: Niños; Fibrosis quística; Signos y síntomas respiratorios; Exacerbacion pulmonar. |
Abstract
|
Cystic fibrosis is the most common autosomal recessive disease among Caucasian populations and is caused by mutations in a single large gene on chromosome 7 that encodes the cystic fibrosis transmembrane conductance regulator (CFTR) protein. The alteration in the CFTR protein results in thickened and viscous secretions in the bronchi, biliary tract, pancreas, intestines, and reproductive system, hence affecting their normal function. The sings and symptoms of lung disease are diverse ranging from asymptomatic infants detected by neonatal screening to older children and adolescents with chronic productive cough, chronic colonization by pathogens such as P. aeruginosa, presence of bronchiectasis and significant alteration of the lung function. Treatment is based on achieving adequate nutrition, reducing bronchial obstruction and promoting mucociliary clearance through respiratory physiotherapy, exercise and early treatment of respiratory infections. Recently, specific therapies that act by improving production, intracellular processing, and/or function of the defective CFTR protein are being incorporated into the treatment, which will revolutionize the prognosis of this disease. Key words: Children; Cystic fibrosis; Signs and symptoms; Respiratory signs and symptoms; Pulmonary exacerbations. |
Introducción
La fibrosis quística (FQ) es la enfermedad genética de herencia autosómica recesiva más frecuente en la población de raza caucásica. Se calcula una prevalencia general de aproximadamente 1 por cada 1.800 – 25.000 recién nacidos vivos, que varía dependiendo de la etnia y la región(1). La incidencia es mayor en Europa, Norteamérica y Australia (1/3.000) y menor en Sudamérica (1/7.000), Asia (1/350.000) y África (1/12.000)(2).
La fibrosis quística (FQ) es la enfermedad genética de herencia autosómica recesiva más frecuente en la población de raza caucásica
En España, se estima una incidencia de fibrosis quística de aproximadamente uno por cada 5.000-10.000 recién nacidos vivos(1). Desde el inicio del cribado neonatal en 2009, se cuenta con cifras exactas de algunas comunidades autónomas, pero no se cuenta con datos publicados de la comunidad de Madrid o datos generales a nivel nacional.
Genética
La fibrosis quística se produce como resultado de la mutación en el gen CFTR (gen regulador de la conductancia transmembrana de la fibrosis quística). Este gen fue aislado y clonado en el año 1989, y se encuentra localizado en el brazo largo del cromosoma 7. Consta de 250 kb, distribuidos en 27 exones y codifica una proteína de 1480 aminoácidos, la proteína CFTR(3).
Se produce como resultado de la mutación en el gen CFTR (gen regulador de la conductancia transmembrana de la fibrosis quística) que se encuentra localizado en el brazo largo del cromosoma 7
Esta proteína está situada en la membrana apical de las células epiteliales de diferentes órganos y sistemas y se encarga de regular los canales de transporte de cloro y sodio y de la activación de otros canales secundarios que participan en el transporte de electrólitos. El transporte anormal de iones, ocasiona el aumento de la viscosidad de las secreciones dando origen a las distintas manifestaciones respiratorias (afectación pulmonar progresiva), digestivas (insuficiencia pancreática, afectación hepática), deshidratación hiponatrémica por pérdida de iones en el sudor o a la infertilidad en los varones por azoospermia obstructiva(4).
Hasta la fecha, se han descrito casi 2.000 mutaciones productoras de fibrosis quística, registradas en el Cystic Fibrosis Database (la base de datos más completa sobre genética en FQ). De estas, 40% originan la sustitución de un aminoácido, 36% alteran el procesamiento del RNA, 3% involucran grandes reordenamientos en la secuencia de CFTR, 1% afectan distintas regiones del promotor, 14% son variantes neutrales y 6% continúan siendo desconocidas.
De manera clásica las mutaciones productoras de FQ, se clasifican en 6 clases funcionales:
- Las mutaciones de clase I (error en la producción de la proteína), impiden la traducción completa de la proteína CFTR debido a un condón de parada prematuro, originando una proteína más corta y no funcionante.
- Las de clase II (error en el procesamiento de la proteína), codifican una proteína mal plegada, estructuralmente alterada y no funcionante, que finalmente es eliminada por el retículo endoplasmático. En este grupo se encuentra la mutación F508del.
- En las de clase III a VI, las proteínas llegan a la superficie de la célula pero no funcionan de manera adecuada. Las de clase III (defecto en la regulación), tienen disminuida la activación del canal y permanecen cerradas. Las de clase IV (defecto en la conducción), tienen alterada la conductibilidad de iones a través del canal. Las de clase V (menor cantidad de proteína funcionante), codifican menor cantidad de proteína, con lo cual, su función se ve reducida y finalmente las de clase VI, codifican una proteína alterada con menor vida media(2).
Las mutaciones de clase I, II y III se conocen como “mutaciones graves”, ya que se asocian con formas clínicas más graves y el resto de grupos se asocia a formas clínicas más leves.
Diagnóstico
Según las manifestaciones clínicas, se debe sospechar de fibrosis quística ante la presencia de enfermedad respiratoria sugestiva, alteraciones digestivas, deshidratación hiponatrémica por pérdida de sales en el sudor o azoospermia secundaria a ausencia bilateral de conductos deferentes.
Cribado neonatal
Dada la importante morbimortalidad de esta enfermedad, muchos países cuentan con estrategias de cribado neonatal para su detección precoz.
En España, el cribado neonatal se instauró en todas las comunidades autonómas entre 1999 y 2015. No obstante, cada comunidad autónoma ha definido su propia estrategia de cribado.
El cribado neonatal se inicia con la determinación de tripsina inmunorreactiva (TIR) en una muestra de sangre de talón, entre el tercer y quinto día de vida. Si la primera TIR es normal, se considera que el resultado del cribado es negativo y se informa a la familia. De lo contrario, se continúa el estudio(5).
En Madrid, el cribado neonatal se inició en junio del 2009 y se realiza bajo la estrategia TIR/DNA. Se continúa el estudio, con una segunda determinación de TIR entre los 25-40 días de vida (se considera positivos valores de TIR por encima de 35 mg/dl). Si los valores de TIR son normales, se considera el cribado negativo. Si continúa elevada, se realiza el estudio genético en la misma muestra de sangre y se envía al paciente a una unidad especializada para la realización de la prueba del sudor(1).
Prueba del sudor
A pesar de la extensión del cribado neonatal, esta prueba sigue siendo la piedra angular del diagnóstico de FQ. Se basa en la medición de los niveles de cloro en sudor, poniendo así de manifiesto, la disfunción de la proteína CFTR.
Está indicada en recién nacidos con cribado neonatal positivo, niños con síntomas sugestivos de FQ y gemelos de niños con FQ, cuando el diagnóstico no se ha podido realizar por estudio genético.
En recién nacidos con cribado positivo, se debe esperar hasta las dos semanas de vida o hasta que tengan un peso por encima de 2 kilos para obtener un resultado óptimo.
El procedimiento se inicia con la estimulación de la sudoración, mediante iontoforesis con pilocarpina y la recogida del sudor con un papel de filtro o gasa previamente pesada, o mediante el sistema Macroduct y posteriormente, se determina la cantidad de cloro empleando un clorhidrómetro.
Un resultado de 60 mmol/l o superior se considera positivo, mientras que valores entre 30 y 59 mmol/l se consideran dudosos. Entre el 1-2% de los pacientes con FQ pueden dar resultados normales o dudosos en especial aquellos portadores de algunas mutaciones como c.3717+12191 C>T(6).
Estudio genético
El hallazgo de dos mutaciones productoras de FQ en ambas copias del gen CFTR confirma el diagnóstico. No todas las mutaciones descritas hasta el momento, se correlacionan claramente con la producción de enfermedad.
Tras el estudio diagnóstico, se puede establecer tres escenarios:
Fibrosis quística clásica: paciente con rasgos fenotípicos, prueba del sudor positiva y dos mutaciones genéticas productoras de FQ.
Fibrosis quística no clásica: paciente con rasgos fenotípicos, prueba del sudor dudosa o normal y dos mutaciones productoras de FQ.
Fibrosis quística de significado incierto o no concluyente (CFSPID): recién nacido con cribado neonatal positivo, prueba del sudor dudosa y una mutación productora de FQ o ninguna mutación o recién nacido con cribado neonatal positivo, prueba del sudor normal y al menos una mutación sin expresión fenotípica clara(6).
Manifestaciones clínicas de la enfermedad pulmonar
En las últimas décadas, la FQ ha pasado de ser una enfermedad pediátrica, con afectación digestivo-nutricional y respiratoria, a ser una enfermedad de adolescentes e incluso adultos, compleja y multisistémica.
La FQ ha pasado de ser una enfermedad pediátrica, con afectación digestivo-nutricional y respiratoria, a ser una enfermedad de adolescentes e incluso adultos, compleja y multisistémica
El espectro clínico de la enfermedad pulmonar es muy amplio y abarca desde lactantes asintomáticos, diagnosticados mediante cribado neonatal o por síntomas clínicos extrapulmonares (gastrointestinales, deshidratación, ileo meconial), a niños mayores e incluso adultos con síntomas respiratorios crónicos de larga evolución y enfermedad pulmonar avanzada.
Desde el inicio del cribado neonatal en España, la mayor parte de los pacientes con FQ son diagnosticados al nacimiento, cuando aún se encuentran asintomáticos. Durante la etapa neonatal, la enfermedad se puede presentar como obstrucción intestinal secundaria a ileo meconial o ictericia, siendo muy poco frecuente la presentación con clínica respiratoria.
Durante la lactancia y la etapa preescolar, algunos pacientes pueden debutar con síntomas respiratorios precoces, a menudo, en forma de bronquiolitis de tórpida evolución. A lo largo de la infancia, puede manifestarse también como neumonías de repetición acompañadas de signos de hiperinsuflación pulmonar o asma grave de mala evolución.
El espectro clínico de la enfermedad pulmonar es muy amplio y abarca desde lactantes asintomáticos, diagnosticados mediante cribado neonatal o por síntomas clínicos extrapulmonares a niños mayores e incluso adultos con síntomas respiratorios crónicos de larga evolución y enfermedad pulmonar avanzada
Durante la adolescencia y la juventud, los síntomas respiratorios se hacen más llamativos. Se caracterizan por la aparición de tos productiva con esputos amarillentos, verdosos y viscosos, colonización de las secreciones respiratorias por distintos patógenos, aparición de bronquiectasias y alteración de la función pulmonar.
Durante la adolescencia y la juventud, los síntomas respiratorios se hacen más llamativos. Se caracterizan por la aparición de tos productiva con esputos amarillentos, verdosos y viscosos, colonización de las secreciones respiratorias por distintos patógenos, aparición de bronquiectasias y alteración de la función pulmonar
Otra manifestación común es la nasosinusal, caracterizada por la aparición de poliposis nasal que afecta a más del 50% de pacientes con FQ y algunas veces puede asociar mucoceles o abscesos periorbitarios. Se presenta como tos, obstrucción nasal, anosmia, respiración bucal y rinorrea en niños o cefalea en adolescentes y adultos(7).
La exploración pulmonar suele ser normal durante años. A medida que la enfermedad avanza, se puede apreciar taquipnea y distintas anomalías auscultatorias como crepitantes, roncus, sibilantes o hipoventilación alveolar. Además, se puede encontrar malformaciones torácicas como cifosis o tórax en quilla. De igual manera, algunos pacientes, con mayor gravedad de la enfermedad, pueden presentar acropaquias.
La aparición de síntomas respiratorios persistentes se asocia a cambios en la función pulmonar y las imágenes radiológicas. En fases iniciales, la obstrucción de la vía aérea pequeña ocasiona alteración en el patrón de ventilación/perfusión, atrapamiento aéreo y atelectasias, manifestándose con un patrón obstructivo en la espirometria forzada. En fases más avanzadas, se observa un patrón mixto con atrapamiento aéreo. Asimismo, hasta el 50% de los pacientes con FQ asocia hiperreactividad bronquial con prueba broncodilatadora positiva.
En cuanto a las imágenes radiológicas, al inicio de la enfermedad, se observa en la radiografía de tórax hiperinsuflación con aplanamiento de las cúpulas diafragmáticas y engrosamiento peribronquial. A medida que la afectación pulmonar empeora, se detecta imágenes nodulares y micronodulares por impactación mucosa en los bronquios, bronquiectasias, bullas subpleurales y finalmente condensaciones multifocales e imágenes cavitadas que representan grandes bronquiectasias con nivel aéreo. En la tomografía torácica, se puede observar cambios más precoces, incluso aun en lactantes y niños pequeños asintomáticos(8).
Progresión de la enfermedad pulmonar
La obstrucción crónica ocasionada por las secreciones viscosas propias de la enfermedad, condiciona la colonización de la vía aérea por distintas bacterias entre las que se puede destacar: Haemophilus influenzae, Staphylococcus aureus, Pseudomonas aeruginosa y el complejo Burkholderia cepacia. En algunas regiones, también es frecuente encontrar cepas de S. aureus resistente a meticilina (MRSA).
Otras bacterias encontradas con frecuencia en estos pacientes son: Stenotrophomonas maltophilia, Achromobacter xylosoxidans y Klebsiella sp. aunque la contribución patogénica de estos microorganismos no está clara.
Estos microorganismos, presentan una secuencia temporal relativamente establecida y asociada a la edad del paciente con FQ. Incluso en aquellos pacientes asintomáticos, detectados por cribado neonatal, se puede evidenciar la presencia de microorganismos en las secreciones respiratorias durante los primeros meses de vida.
Durante las primeras etapas de la vida, las infecciones víricas propias de la infancia provocan la denudación del epitelio pulmonar, favoreciendo así la colonización bacteriana recurrente y el estado local de inflamación crónica. Se ha demostrado, que algunos virus como el Adenovirus y Coronavirus y también determinadas bacterias como Micoplasma pneumoniae y Chlamydophila pneumoniae estimulan el sistema fagocítico, favoreciendo la descamación del epitelio y la atracción de neutrófilos.
Tras el periodo inicial, los patógenos colonizadores de la vía aérea más frecuentes son S. aureus y Haemophylus influenzae. S. aureus suele ser el patógeno predominante durante esta etapa y el que usualmente inicia el periodo de colonización. Streptococccus pneumoniae también se encuentra con frecuencia en la vía aérea de estos pacientes, pero no tiene una incidencia mayor que en niños sanos. No obstante, si suele tener un patrón de resistencia mayor(9).
De manera que la edad del paciente va incrementándose, disminuye la colonización por S. aureus y aumenta el aislamiento de P. aeruginosa. Esta bacteria se incrementa de forma gradual en las secreciones respiratorias hasta convertirse en el patógeno más frecuentemente aislado en la edad adulta. Se le ha asociado al deterioro progresivo e irreversible de la función pulmonar y se le considera la causa más importante de morbimortalidad pulmonar. (Figura 1).
La hiperviscosidad de las secreciones asociada al estado proinflamatorio de la vía aérea genera un ambiente óptimo para la aparición de cualidades fenotípicas específicas en P. aeruginosa. La hipoxia generada por este estado, incentiva la producción de alginato y la disminución de motilidad de la bacteria, propiciando la formación de grandes macrocolonias bacterianas (biofilms), en las cuales, las bacterias permanecen en un estado quiescente y de baja replicación, ocasionando que su erradicación sea casi imposible(10).
En adolescentes o pacientes que han recibido muchos ciclos antibióticos, no es raro encontrar también patógenos como Aspergillus fumigatus, distintas especies de Candida sp o micobacterias atípicas. A. fumigatus se relaciona con la aparición de aspergilosis broncopulmonar alérgica en tanto Candida sp. se considera un patógeno saprofito sin relevancia clínica. En cuanto a las micobacterias atípicas, la que se aísla con más frecuencia suele ser Mycobacterium avium.
En la mayoría de los pacientes, casi el 70%, el patrón de colonización respiratoria suele ser polimicrobiano coexistiendo distintos patógenos al mismo tiempo(9).
Exacerbación pulmonar
La FQ condiciona un estado crónico de inflamación, obstrucción e infección de la vía aérea. A esto, se suma la aparición de numerosos episodios de exacerbación pulmonar o reagudización infecciosa que aceleran el deterioro de la función pulmonar.
No existen criterios claros para definir la exacerbación pulmonar. De manera clásica, se utilizaban los criterios definidos por Fusch y cols. Actualmente, el Euro Care CF Working Group busca validar los criterios de Fusch y cols modificados, que incluyen cambios que los hacen más adecuados para la práctica clínica habitual (Tabla I)(11).
El número de exacerbaciones por año se correlaciona con la disminución de la función pulmonar independiente de la edad del paciente y del tratamiento administrado. Por este motivo, es muy importante la detección precoz y el correcto tratamiento de las exacerbaciones pulmonares.
En pacientes mayores de 18 años se observa una relación lineal entre el número de exacerbaciones y el grado de deterioro de la función pulmonar (expresado como un FEV1 bajo), es decir a menor función pulmonar mayor número de exacerbaciones pulmonares. Por otro lado, en pacientes menores de esta edad, la correlación es exponencial.
Existen factores de riesgo que predisponen a un mayor número de exacerbaciones pulmonares como: el sexo femenino, la malnutrición, la insuficiencia pancreática, la infección persistente por S. aeruginosa, B. cepacea o MRSA, la aspergilosis broncopulmonar, un gran descenso del FEV1 y las infecciones virales(12).
Las exacerbaciones pulmonares se pueden clasificar en:
Exacerbación leve-moderada: sintomatología leve que no afecta el estado general, la tolerancia al ejercicio ni las actividades diarias del paciente.
Exacerbación grave: se asocian a afectación importante del estado general, disnea a mínimos esfuerzos u ortopnea o hipoxemia. Precisan en la mayoría de las ocasiones de tratamiento intravenoso.
Pequeñas recaídas infecciosas pueden descompensar llamativamente a un paciente con enfermedad pulmonar avanzada, mientras que infecciones importantes pueden perturbar escasamente a otro con una enfermedad inicial(13).
La adolescencia y la pubertad juegan un rol importante en la evolución de la enfermedad. A partir de la adolescencia, se observa un mayor número de exacerbaciones y de mayor gravedad, en mujeres respecto a hombres, así como una mayor incidencia de colonización por P. aeruginosa. Esta evolución dispar, estaría relacionada con el papel que juegan las hormonas sexuales femeninas (estrógenos y progestágenos) sobre la función pulmonar(14).
A partir de la adolescencia, se observa un mayor número de exacerbaciones y de mayor gravedad, en mujeres respecto a hombres, así como una mayor incidencia de colonización por P. aeruginosa
El tratamiento de la FQ se basa en lograr una nutrición adecuada, reducir la obstrucción bronquial y promover el aclaramiento mucociliar a través de la fisioterapia respiratoria, el ejercicio y el tratamiento precoz de las infecciones respiratorias
Tratamiento
Aclaramiento mucociliar
Fisioterapia respiratoria: tiene como objetivo movilizar y drenar las secreciones, liberando la vía aérea y con ello disminuye el riesgo de infecciones y mejora la función pulmonar. Se debe iniciar cuanto antes, incluso aun en pacientes asintomáticos.
Ejercicio físico: el ejercicio aérobico aumenta la eliminación de secreciones y mejora los parámetros cardiovasculares.
Tratamiento broncodilatador: solo se recomienda su uso en pacientes con hiperreactividad bronquial o previo a la fisioterapia o a la administración de medicación inhalada(15).
Suero salino hipertónico: la administración de suero salino hipertónico de forma inhalada hidrata las secreciones respiratorias permitiendo un mejor aclaramiento mucociliar. Se postula, que la alta osmolaridad de la solución, estimula el paso de agua hacia el epitelio pulmonar restableciendo la capa acuosa. La administración de suero salino hipertónico a una concentración del 7% en dosis de 4 ml dos veces al día, previo uso de un broncodilatador, ha demostrado ser segura. No obstante, la primera administración de la medicación debe realizarse de manera hospitalaria para vigilar posibles episodios de broncoespasmo(16).
DNasa: la dornasa alfa es una enzima DNasa recombinante que degrada el ADN de los neutrófilos reduciendo la viscosidad de las secreciones purulentas. Se debe administrar de forma inhalada, en una pauta de 2.5 mg una vez al día.
Tratamiento antiinflamatorio
Macrólidos: se ha demostrado que la azitromicina actúa como antiinflamatorio mejorando la función pulmonar y disminuyendo el número de exacerbaciones. Estos efectos, se atribuyen a su capacidad inmunomoduladora que actúa reduciendo el número de neutrófilos y disminuyendo la producción de citoquinas proinflamatorias.
No tiene acción bactericida sobre P. aeruginosa in vitro, aunque se ha observado que actúa a nivel de biofilms, reduciendo la formación de alginato. Se recomienda su uso en mayores de 6 años con infección crónica por P. aeruginosa.
Antes de iniciar su administración, se debe descartar la infección por micobacterias atípicas. Se administra a una dosis de 10 mg/kg en pacientes menores de 40 Kg o 250-500 mg en mayores de dicho peso, a días alternos de manera prolongada.
Ibuprofeno: a dosis altas mejora la función pulmonar y reduce las exacerbaciones respiratorias. Su uso clínico es limitado en la edad pediátrica dado sus efectos adversos.
Corticoides sistémicos: no se recomienda su uso en pacientes pediátricos salvo en casos de asma o aspergilosis broncopulmonar alérgica(15).
Moduladores de CFTR
Este grupo de fármacos mejora la producción, procesamiento intracelular y la función de la proteína CFTR. Las moléculas dirigidas a corregir las mutaciones de clase I, II, VI se denominan correctoras del CFTR y las dirigidas a que la proteína mejore su función se denominan potenciadoras (grupos III, IV, V)(2).
Los moduladores de CFTR mejoran la producción, procesamiento intracelular y la función de la proteína CFTR
Ivacaftor: actúa sobre la mutación G551D y en el resto de mutaciones de clase III. Se indica en pacientes mayores de 2 años. En pacientes entre 2-6 años, menores de 14 kilos, se recomienda una dosis de 50 mg cada 12 horas, en tanto en aquellos, mayores de 14 kilos se recomienda 75 mg cada 12 horas. En mayores de 6 años, la dosis recomendada es de un comprimido de 150 mg cada 12 horas(17).
Se debe administrar 30 minutos después de una comida con grasa, para su correcta absorción. La primera dosis del fármaco, debe de ser probada en un medio hospitalario para controlar posibles efectos adversos. Durante el seguimiento clínico se debe controlar las enzimas hepáticas(18).
Lumacaftor-ivacaftor: está indicado en aquellos pacientes homocigotos para la mutación F508del. Lumacaftor corrige parcialmente el mal plegamiento de CFTR e ivacaftor mejora la actividad del canal de Cloro. Se recomienda en mayores de 6 años, a una dosis de 2 tabletas (lumacaftor 200 mg- ivacaftor 125 mg) cada 12 horas.
Tezacaftor- ivacaftor: tezacaftor como lumacaftor mejora el procesamiento intracelular de CFTR. Su uso está indicado en homocigotos de F508del mayores de 12 años. Se recomienda el tratamiento con una dosis de tezacaftor 100 mg/ivacafotr 150 mg por la mañana e ivacaftor 150 mg por la noche(2).
Tratamiento de las exacerbaciones pulmonares
El tratamiento de las exacerbaciones pulmonares debe realizarse según los resultados del cultivo y el antibiograma de las muestras de secreciones respiratorias previas. Debe tenerse en cuenta la respuesta terapéutica en ocasiones anteriores.
Se debe tratar cualquier exacerbación debida a especies de P. aeruginosa y S. aureus, así como a especies de Achromobacter xylosoxidans, dada la asociación que existe con tasas de deterioro del FEV1 (similares a las inducidas por P. aeruginosa)(19).
Existe más incertidumbre en cuanto a la importancia de tratar la infección por Stenotrophomonas maltophilia. En algunos pacientes, se ha observado un claro deterioro clínico siendo S. maltophilia el único agente patógeno presente en las muestras respiratorias.
El tratamiento antibiótico puede realizarse de forma oral o intravenosa. No obstante, en exacerbaciones graves o en caso de falla del tratamiento oral, se debe administrar la medicación por vía intravenosa.
En la mayoría de los casos, se puede administrar la medicación intravenosa de manera domiciliaria mediante el uso de catéteres centrales de canalización periférica (PICC). Sin embargo, en caso de exacerbaciones graves con insuficiencia respiratoria que precisen de oxigenoterapia continua, en caso de pacientes con complicaciones como una hemoptisis significativa; broncorrea cuantiosa que requiera de un apoyo intensivo de fisioterapia; pacientes con insuficiencia renal o diabetes descompensada o cuando sea imposible asegurar una cumplimentación cuidadosa en el domicilio se debe administrar la medicación en el medio hospitalario(11).
La duración del tratamiento es de 14 a 21 días. En caso de infección por P.aeruginosa y B. cepacea se recomienda el tratamiento combinado con dos antibióticos de administración sistémica (Tabla II).
De igual manera, en caso de P. aeruginosa se debe agregar tratamiento con fármacos inhalados como tobramicina cada 12 horas (300 mg/5ml) en ciclos de 28 días on y 28 dias off, colistimetato de sodio en dosis de 1-2 millones de unidades cada 12 horas o aztreonam 75 mg cada 8 horas(10).
Junto al tratamiento antibiótico, se deben intensificar el tratamiento nutricional, la terapia inhalada y la fisioterapia para obtener mejores resultados.
Tablas y figuras
Tabla I. Definición de exacerbación pulmonar
|
EURO CARE CF WORKING GROUP 2011 |
|
El grupo del consenso europeo desea validar los criterios modificados de Fuchs y cols: Una exacerbación será definida como la necesidad de tratamiento antibiótico adicional indicado por un cambio reciente de al menos dos de los siguientes:
|
Tabla II. Tratamiento de las exacerbaciones pulmonares
|
EXACERBACIONES LEVES |
|
S. aureus sensible a meticilina, H. influenzae: Amoxicilina-acido clavulánico 80-90 mg/Kg/día cada 8 horas VO durante 14 días. MRSA, B.cepacea, S. maltophilia: CotrimoxazolVO 400 mg/Kg/día cada 6 horas durante 14 días. P. aeruginosa: Ciprofloxacino 40 mg/Kg/día cada 12 horas VO durante 14 días +/- colistimetato sódico inhalado (Promixin) 2 MU cada 8 horas. |
|
EXACERBACIONES GRAVES |
|
S. aureus sensible a meticilina, H. influenzae: Amoxicilina-acido clavulánico 100 mg/Kg/día cada 8 horas IV durante 14 días. P. aeruginosa: Ceftazidima 150 mg/Kg/día cada 8 horas + Tobramicina 10 mg/Kg/día cada 24 horas durante 14 días +/- colistimetato sódico inhalado (Promixin) 2 MU cada 8 horas. MRSA, Vancomicina IV 60 mg/Kg/día cada 6 horas durante 14 días. S. maltophilia, B.cepacea: alta resistencia valorar según antibiograma. OPCIÓN: Piperacilina 400 mg/Kg/día cada 6 horas + tobramicina 10 mg/Kg/día cada 24 horas. |
Figura 1. Colonización bacteriana según edad
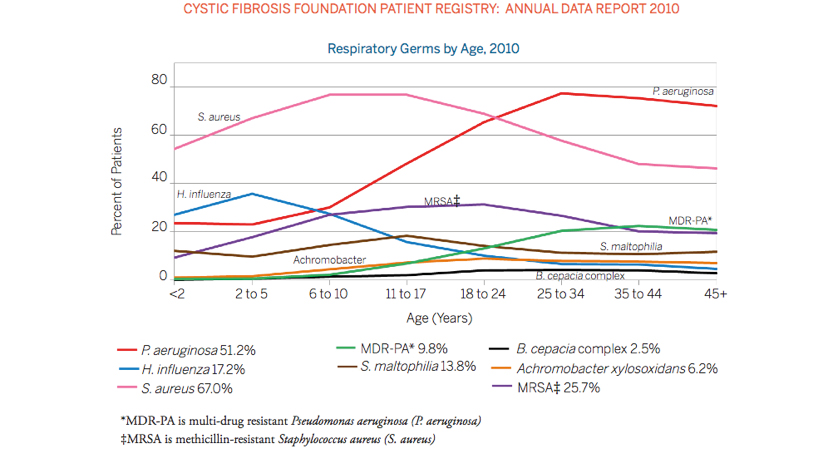
Fuente: Cystic Fbrosis Foundation Patient Registry 2010.
Bibliografía
- Gartner S, Cobos N. Cribado neonatal para la fibrosis quística. An Pediatr (Barc). 2009; 71: 481-2.
- Quon B, Rowe S. New and emerging targeted therapies for cystic fibrosis. BMJ. 2016;352: i859.
- Koch C, N Hoiby. Pathogenesis of cystic fibrosis. Lancet. 1993; 341: 1965-9.
- Schwiebert EM, Benos DJ, Egan ME, Atutts MJ, Guggino WB. CFTR is a conductance regulator as well as a chloride cannel. Physiol Rev. 1999; 79: S145-66.
- Castellani C, Southern KW, Brownlee K, Dankert J, Duff A, Farrel M et al. European best practices guidelines for cystic fibrosis neonatal screening. J Cyst Fibros. 2009; 8: 153-73.
- Farrell P, White T, Ren C, Hempstead S, Accurso F, Derich N et al. Diagnosis of cystic fibrosis: Consensus Guidelines from the Cystic Fibrosis Foundation. J Pediatr. 2017; 181S:4-15.
- Gartner S, Salcedo A, Garcia G. Enfermedad respiratoria en la fibrosis quística. Protoc diagn ter pediatr. 2017; 1:299-319.
- Hall GL, Logie KM, Parsons F et al. Air trapping on Chest TC is associated with worse ventilation distribution in infants with cystic fibrosis diagnosed following enwborn screening. Plos One. 2011; 6: e23932.
- De Vrankriijker A, Wolfs T, Van Der Ent C. Challenging and emerging patrohgens in cystic fibrosis: Paediatr Respr Rev 2010; 11: 246-54.
- Cantón R, Maiz L, Escribano A et al. Consenso español para la prevención y el tratamiento de la infección bronquial por Pseudomonas aeruginosa en el paciente con fibrosis quística. Arch Bronconeumol. 2015; 51: 140-50.
- Bilton D, Canny G et al. Pulmonary exacerbations: towards a definition for use in clinical trials. Journal of cystic fibrosis 2011; 10: 79-81.
- Goss C, Burns J. Exacerbations in cystic fibrosis: epidemiology and pathogenesis. Thorax 2007; 62:360-367.
- Flume P, Mogayzel P et al. Cystic fibrosis pulmonary guidelines. Am J Respir Crit Care Med 2009; 180: 802-808.
- Sutton S, Rosenbluth D, Raghavan D, Zhebg J et al. Effects of puberty on cystic fibrosis related pulmonary exacerbations in women versus men. Pediatr Pulmonol 2014: 49(1): 28-35.
- Davis PB. Therapy for cystic fibrosis–the end of the beginning? N Engl J Med 2011; 365:1734.
- Elkins MR, Robinson M, Rose BR et al. A controlled trial of long-term inhaled hypertonic saline in patients with cystic fibrosis. N Engl J Med. 2006; 354:229-40.
- Lahiri T, Hempstead SE, Brady C, et al. Clinical Practice Guidelines From the Cystic Fibrosis Foundation for Preschoolers With Cystic Fibrosis. Pediatrics 2016; 137.
- Davies JC, Wainwright CE, Canny GJ, et al. Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. Am J Respir Crit Care Med 2013; 187:1219. Ramsey BW, Davies J, McElvaney NG, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med 2011; 365:1663.
- Bhatt J. Treatment of pulmonary exacerbations in cystic fibrosis. Eur Respir Rev 2013; 22:205-216.

