Conectivopatías en adolescentes
Conectivopatías en adolescentes
Daniel Clemente Garulo. Juan Carlos López Robledillo.
Unidad de Reumatología Pediátrica. Hospital Infantil Universitario Niño Jesús. Madrid.
Fecha de recepción: 11 de noviembre 2014
Fecha de publicación: 15 de febrero 2015
Adolescere 2015; III (1): 30-43
Resumen
|
Las conectivopatías constituyen un grupo heterogéneo de enfermedades ocasionadas por el desarrollo de autoanticuerpos o células T autorreactivas frente a diversas estructuras corporales. Se caracterizan por una afectación multisistémica sin una causa aparente, con síntomas y signos variables que pueden aparecer simultáneamente o durante el transcurso de semanas o meses. Es habitual la presencia de anticuerpos antinucleares y otros autoanticuerpos y ayuda a la confirmación diagnóstica. Pueden ser enfermedades graves y los tratamientos empleados para controlar la enfermedad, principalmente corticoides e inmunosupresores, pueden dar lugar a efectos adversos significativos a corto y largo plazo.. Palabras clave: conectivopatías, lupus eritematoso sistémico, dermatomiositis juvenil, esclerodermia pediátrica. |
Abstract
|
Connective tissue diseases are a heterogeneous group of disorders caused by the development of autoantibodies or autoreactive T cells against various body structures. They are characterized by multi-system involvement without apparent cause, with variables signs and symptoms that may appear simultaneously or in the course of several weeks and months. It is common to find anti-nuclear and other autoantibodies and helps confirm the diagnosis. These diseases can be severe and the treatments required for disease control, mainly steroids and immunosuppressive drugs, have potential side effects in the short and long term. Key words: connective tissue diseases, systemic lupus erythematosus, juvenile dermatomyositis, pediatric scleroderma. |
Introducción
Las conectivopatías constituyen un grupo heterogéneo de enfermedades ocasionadas por una alteración en la inmunidad adquirida, apareciendo autoanticuerpos o células T autorreactivas frente a diversas estructuras corporales. La causa es desconocida, aunque en su aparición va a influir la combinación de una serie de factores ambientales en un paciente predispuesto genéticamente. Aunque son poco frecuentes en adolescentes, es importante su reconocimiento precoz para iniciar un tratamiento adecuado que permita el control de la enfermedad y mejorar el pronóstico de los pacientes a largo plazo.
Prevalencia
Las conectivopatías son enfermedades raras, con un predominio del sexo femenino y tendiendo a aparecer en adolescentes o preadolescentes
Las conectivopatías son enfermedades raras, con un predominio del sexo femenino y tendiendo a aparecer en adolescentes o preadolescentes. El lupus eritematoso sistémico es la conectivopatía más conocida, con una incidencia de 0,3 a 0,9 casos/100.000 niños/año y una prevalencia de 3,3 a 8,8/100.000 niños, afectando especialmente a asiáticos, afroamericanos y latinos. Otras conectivopatías son la dermatomiositis juvenil (2-4 casos/millón de niños), la esclerodermias localizadas (1 caso por cada 100.000/niños), la esclerosis sistémica (1 caso por cada millón de niños), la enfermedad mixta del tejido conectivo y el síndrome de Sjögren.
Manifestaciones clínicas
Las conectivopatías se caracterizan por afectar a múltiples órganos y sistemas sin una causa aparente, con síntomas y signos variables
Las conectivopatías se caracterizan por afectar a múltiples órganos y sistemas sin una causa aparente, con síntomas y signos variables que pueden aparecer simultáneamente o de forma insidiosa durante el transcurso de semanas o meses. Los síntomas iniciales son inespecíficos en muchos casos, como fiebre, astenia, anorexia, pérdida de peso o linfadenopatías. Por ello debe establecerse un diagnóstico diferencial con infecciones (víricas y bacterianas), neoplasias (especialmente procesos linfoproliferativos) y otras enfermedades inflamatorias (vasculitis). Las manifestaciones clínicas van a depender de los órganos afectados, siendo algunas de ellas muy sugerentes de una conectivopatía (exantema malar en lupus eritematoso o pápulas de Gottron en dermatomiositis juvenil).
Lupus eritematoso sistémico
El lupus eritematoso sistémico (LES) es una enfermedad autoinmune provocada por la aparición de autoanticuerpos dirigidos frente a múltiples órganos y sistemas
El lupus eritematoso sistémico (LES) es una enfermedad autoinmune provocada por la aparición de autoanticuerpos dirigidos frente a múltiples órganos y sistemas. Los síntomas constitucionales, la artritis y el exantema malar son las manifestaciones iniciales más frecuentes en los pacientes con LES juvenil. Durante su evolución puede afectarse cualquier órgano, apareciendo de forma aislada o simultáneamente manifestaciones mucocutáneas (exantema malar en alas de mariposa (imagen 1), fotosensibilidad, úlceras orales, alopecia, fenómeno de Raynaud), musculoesqueléticas (artritis/artralgias, mialgias, necrosis avascular), renales (HTA, glomerulonefritis), neuropsiquiátricas (cefalea, las alteraciones del estado de ánimo, la psicosis, la disfunción cognitiva, convulsiones), hematológicas (citopenias), gastrointestinales (dolor abdominal, diarrea, hepatoesplenomegalia) y/o cardiopulmonares (pericarditis/pleuritis, alteraciones en las pruebas funcionales respiratorias, miocarditis,…).
Dermatomiositis juvenil
La dermatomiositis juvenil (DMJ) se caracteriza por la inflamación crónica de piel y músculo estriado, presentándose como un cuadro de debilidad muscular
La dermatomiositis juvenil (DMJ) se caracteriza por la inflamación crónica de piel y músculo estriado, presentándose como un cuadro de debilidad muscular de comienzo insidioso y manifestaciones cutáneas específicas como las pápulas de Gottron (imagen 2) o el eritema heliotropo (imagen 3). La debilidad muscular afecta a la cintura escapular (dificultad para levantar los brazos) y pélvica (dificultad para subir y bajar escaleras) y puede asociar dolor muscular con el ejercicio. El signo de Gowers (el niño trepa por sus muslos para levantarse del suelo) es positivo. Puede aparecer disfagia o disnea por afectación de la musculatura faríngea y respiratoria. Durante su evolución pueden aparecer calcinosis, úlceras cutáneas y una lipodistrofia o pérdida de tejido adiposo subcutáneo.
Esclerodermia juvenil
La esclerodermia agrupa a un conjunto de enfermedades caracterizadas por la fibrosis o excesivo depósito de colágeno en la piel y en otros tejidos
La esclerodermia agrupa a un conjunto de enfermedades caracterizadas por la fibrosis o excesivo depósito de colágeno en la piel y en otros tejidos. Dependiendo de la localización y extensión de la fibrosis se distinguen formas localizadas (se afectan piel y tejidos subyacentes) o sistémicas (se afectan piel, vasos y órganos internos). La esclerodermia localizada se inicia de manera insidiosa con un edema cutáneo localizado con bordes eritematosos o violáceos, seguido de un progresivo endurecimiento de la piel y tejidos subyacentes (imagen 4), con frecuencia asociado a alteraciones de la pigmentación, alopecia y anhidrosis. Pueden aparecer manifestaciones extracutáneas, principalmente musculoesqueléticas (artralgias/artritis) y, si se afecta cara y cuero cabelludo, manifestaciones neurológicas (cefalea, convulsiones, trastornos del comportamiento) y oculares (epiescleritis, uveítis, afectación palpebral). La esclerosis sistémica suele iniciarse con la aparición de un fenómeno de Raynaud asociado a edema e induración progresiva en manos y cara, telangiectasias (cara y extremidades superiores) y alteraciones capilares en el lecho ungueal. Las principales manifestaciones extracutáneas son gastrointestinales (reflujo gastroesofágico, trastornos en la motilidad digestiva), respiratorias (alteraciones en las pruebas de función pulmonar, enfermedad intersticial pulmonar, hipertensión pulmonar) y musculoesqueléticas (artralgias y artritis, sobre todo en manos).
Enfermedad mixta del tejido conectivo
La enfermedad mixta del tejido conectivo (EMTC) se caracteriza por presentar signos y síntomas de dos o más conectivopatías y la presencia de los anti-RNP (anticuerpos anti ribonucleoproteína nuclear). Las manifestaciones clínicas iniciales más frecuentes son el fenómeno de Raynaud y la poliartritis de manos, apareciendo progresivamente manifestaciones de artritis idiopática juvenil (sinovitis), LES (exantema malar, pleuritis, pericarditis, adenopatías), DMJ (debilidad muscular, miositis) y esclerosis sistémica (esclerodactilia, enfermedad pulmonar intersticial, trastornos en la motilidad esofágica).
Síndrome de Sjögren
El síndrome de Sjögren se caracteriza por la inflamación de las glándulas exocrinas, principalmente glándulas salivales y lagrimales. Por ello se manifiesta como sequedad oral (dificultad para salivar durante las comidas o para hablar) y ocular además de otros síntomas sistémicos variables.
Pruebas complementarias
Lupus eritematoso sistémico
Aunque los ANA pueden ser positivos en niños sanos, valores por encima de 1/1280 son muy sugerentes de LES
La característica principal del LES es la presencia de múltiples autoanticuerpos, siendo los anticuerpos antinucleares (ANA) positivos (títulos > 1 /160) en la mayoría de pacientes. Aunque los ANA pueden ser positivos en niños sanos, valores por encima de 1/1280 son muy sugerentes de LES. Los anticuerpos anti-DNA (en 75% pacientes) y los anti-Sm (anticuerpos anti Smith)(en 50% pacientes) son muy específicos de LES. Otros anticuerpos que pueden encontrarse son los anti-SS-A (Ro) y anti-SS-B(La), los anti-RNP y los anticuerpos antifosfolípido (anticoagulante lúpico, anticardiolipina). Para monitorizar la actividad de la enfermedad se utilizan los niveles de anti-DNA y los valores de complemento (C3 y C4), siendo estos últimos bajos o indetectables durante los periodos de actividad. El hemograma puede mostrar la citopenia de una o más series hematológicas y puede aparecer elevación de las transaminasas y de la creatinina en el perfil bioquímico. Es habitual la elevación de la VSG con unos valores normales de la PCR, ya que está última suele ser normal durante un brote de LES (excepto si se manifiesta como serositis) y puede ayudar a diferenciar actividad lúpica de infecciones intercurrentes. En el análisis de orina podemos encontrar proteinuria, hematuria y/o cilindros urinarios. En estos casos es necesaria la biopsia renal para establecer el tipo de nefritis lúpica y su actividad (tabla 1).
Dermatomiositis juvenil
La biopsia muscular sigue siendo necesaria para confirmar el diagnóstico en pacientes sin las alteraciones cutáneas características
El hemograma suele ser normal al inicio de la enfermedad, aunque puede aparecer linfopenia. Las enzimas musculares (creatinquinasa, alanina aminotransferasa, aspartato amino transferasa, lactato deshidrogenasa, aldolasa) están elevadas en más del 75 % de los pacientes, pero sus valores no se correlacionan con la actividad de la enfermedad. Los ANA son positivos hasta en el 85% de los casos y pueden aparecer autoanticuerpos específicos de miositis, como el anti-Jo1. La inflamación muscular puede demostrarse mediante un EMG y/o una resonancia magnética, siendo esta última cada vez más utilizada en la evaluación inicial de pacientes con DMJ. o con manifestaciones atípicas (debilidad muscular asimétrica, localizada o de predominio distal, atrofia muscular, fasciculaciones,…). La calcinosis es fácilmente apreciable en las radiografías simples. Es recomendable la realización de una radiografía de tórax y pruebas de función pulmonar al inicio del cuadro y durante su evolución, al igual que un ECG y un ecocardiograma basales. La biopsia muscular sigue siendo necesaria para confirmar el diagnóstico en pacientes sin las alteraciones cutáneas características
Esclerodermia juvenil
En las esclerodermias localizadas no hay alteraciones analíticas características
En las esclerodermias localizadas no hay alteraciones analíticas características, pudiendo los reactantes de fase aguda (VSG, PCR) reflejar la actividad de la enfermedad. Pueden detectarse ANA, aunque sin una correlación clínica significativa, así como FR. En las esclerosis sistémicas el hemograma, la orina y los reactantes de fase aguda suelen ser normales. Casi todos los pacientes tienen ANA, siendo característica la positividad a los anticuerpos antitopoisomerasa (o Scl-70) y los anticuerpos anticentrómero. La radiografía simple detecta la presencia de calcificaciones, erosiones articulares y acrosteolisis. Debe realizarse una radiografía de tórax, pruebas de función pulmonar (espirometría, difusión de la capacidad de monóxido de carbono), un ECG y un ecocardiograma en la valoración inicial de estos pacientes. Cuando existen alteraciones en las pruebas funcionales respiratorias hay que descartar una alveolitis pulmonar mediante un TC de alta resolución y/o un lavado broncoalveolar. Ante síntomas de reflujo gastroesofágico debe completarse el estudio con una manometría, una pHmetría y/o una endoscopia digestiva alta.
Enfermedad mixta del tejido conectivo
Los títulos de ANA están elevados, siendo característica la presencia de anticuerpos anti-RNP a títulos altos
Es frecuente encontrar en el hemograma anemia, leucopenia y/o trombocitopenia. Otras alteraciones comunes son la hipocomplementemia, la elevación de las enzimas musculares y la presencia de FR positivo. Los títulos de ANA están elevados, siendo característica la presencia de anticuerpos anti-RNP a títulos altos.
Síndrome de Sjögren
La presencia de ANA y anticuerpos anti-SSA o anti-SSB refuerza la sospecha diagnóstica, que puede confirmarse mediante el estudio anatomopatológico de biopsias de glándulas salivares menores.
Diagnóstico
Se han establecido unos criterios para el diagnóstico de lupus eritematoso sistémico (tabla 1), dermatomiositis juvenil (tabla 2) y esclerosis sistémica (tabla 3). En el resto de las conectivopatías el diagnóstico se realiza principalmente mediante la historia clínica y la exploración física y con el apoyo de las pruebas complementarias.
Tratamiento
El tratamiento de las conectivopatías se basa en el uso de fármacos, principalmente corticoides e inmunosupresores, y en la atención a medidas generales de salud. Es fundamental la educación del adolescente y la familia, ya que el cumplimiento del tratamiento a largo plazo puede ser difícil, bien por miedo a efectos adversos (estrías cutáneas o aumento de peso con altas dosis de corticoides) o porque se “cansen” de estar enfermos, acudir a citas, tomar medicaciones,… Hay que asegurar que el calendario vacunal ha sido completado (especialmente la inmunización contra el neumococo) y realizar un tratamiento precoz de las infecciones.
Lupus eritematoso sistémico
Debe evitarse la exposición al sol (radiación UVB) en adolescentes con LES, recomendando protección solar (SPF ≥ 30) a diario
Debe evitarse la exposición al sol (radiación UVB) en adolescentes con LES, recomendando protección solar (SPF ≥ 30) a diario. Por supuesto, no se recomienda broncearse ni acudir a salones de bronceado. Se debe recomendar una ingesta adecuada de calcio y suplementos de vitamina D y controlar la dieta cuando se inicia un tratamiento con corticoides a dosis altas para evitar una excesiva ganancia ponderal. Especialmente los adolescentes con LES deben evitar fumar, ya que puede empeorar la actividad de la enfermedad y disminuir la eficacia de medicaciones como la hidroxicloroquina.
El tratamiento farmacológico del LES debe ser individualizado según las manifestaciones clínicas asociadas, la extensión y la gravedad de la enfermedad. Los fármacos más habituales y sus indicaciones se recogen en la tabla 5. La dosis y duración del tratamiento con corticoides está basada en la gravedad de las manifestaciones clínicas, desde una dosis de prednisona de 0.25-0.75 mg/kg/día en dosis única (manifestaciones cutáneas y articulares, serositis y síntomas sistémicos generales) a dosis iniciales de 2 mg/kg/día repartida en 3 dosis cuando existe afectación renal grave (nefritis lúpica III o IV) o manifestaciones neuropsiquiátricas. La administración temprana de bolos de metilprednisolona de 30 mg/kg vía intravenosa (3 días consecutivos) debe considerarse en estos casos graves. Con dosis altas de corticoides la aparición de efectos adversos, como acné, vello facial (transitorios) o estrías cutáneas (permanentes) es prácticamente constante, pudiendo aparecer necrosis avascular y mayor número de infecciones. El tratamiento con ciclofosfamida en menores de 20 años se asocia a fallo ovárico en un 13 % de los casos, por lo que puede plantearse el uso de análogos de hormona liberadora de gonadotropina como medida preventiva.
Dermatomiositis juvenil
El tratamiento de la DMJ se basa en la administración de metotrexato (15 mg/m2/semana) asociado a prednisona a dosis de 2 mg/kg/día vía oral con un descenso progresivo
El tratamiento se basa en la administración de metotrexato (15 mg/m2/semana) asociado a prednisona a dosis de 2 mg/kg/día vía oral con un descenso progresivo a las 2-4 semanas hasta su suspensión 12-24 meses después. Como terapia adyuvante de las lesiones cutáneas puede utilizarse tratamientos tópicos (corticoides, tacrólimos, pimecrolimus) o asociar hidroxicloroquina oral a 3-6 mg/kg/día. También se recomiendan suplementos de calcio y vitamina D y evitar la exposición solar. Aunque en la fase aguda se recomienda el reposo, el ejercicio físico aeróbico moderado aporta beneficios a los pacientes en remisión clínica.
Esclerodermia localizada
En los casos en los que existe afectación profunda de la esclerodermia localizada, se utilizan corticoides orales (prednisona a 0,5-1 g/kg/día) asociado a metotrexato a dosis de 10-15 mg/m2/semana vía oral o subcutánea
En las lesiones circunscritas superficiales con signos de actividad puede realizarse tratamiento tópico con corticoides, inhibidores de la calcineurina (tacrólimus, pimecrolimus), o con fototerapia con luz ultravioleta (UV). En los casos en los que existe afectación profunda, que cruza articulaciones (riesgo de limitación funcional), lesiones lineales o atróficas que afectan a cara o cuero cabelludo, una progresión rápida o una distribución amplia de las lesiones y/o un fracaso a los tratamiento tópicos o terapia UV, se utilizan corticoides orales (prednisona a 0,5-1 g/kg/día) asociado a metotrexato a dosis de 10-15 mg/m2/semana vía oral o subcutánea. Puede ser necesaria la realización de fisioterapia para mejorar contracturas articulares y cirugía plástica para la reconstrucción de alteraciones faciales.
Esclerosis sistémica
El tratamiento de la esclerosis sistémica es sintomático
El tratamiento de la esclerosis sistémica es sintomático y depende de las manifestaciones clínicas del paciente, como el nifedipino para el fenómeno de Raynaud o inhibidores de la bomba de protones y procinéticos para el reflujo gastroesofágico. Algunas recomendaciones generales incluyen una protección contra el frío y los traumatismos y la adecuada hidratación de la piel. Se necesitan programas de fisioterapia para mejorar las contracturas articulares.
Enfermedad mixta del tejido conectivo
La mayoría de pacientes suelen responder a corticoides a dosis bajas, AINEs, hidroxicloroquina o combinaciones de estas medicaciones. El fenómeno de Raynaud se trata evitando el frio y el estrés emocional y, en los casos más graves, con nifedipino. Cuando existe una afectación visceral importante se requieren corticoides a dosis altas e inmunosupresores.
Síndrome de Sjögren
El tratamiento suele ser sintomático: lágrimas artificiales, estimulantes de la salivación, una buena higiene dental y antiinflamatorios no esteroideos para los dolores articulares.
Evolución y pronóstico
El LES en niños y adolescentes tiene peor pronóstico que los casos de inicio en la edad adulta
Las conectivopatías son enfermedades crónicas que suelen cursar con periodos de actividad y de remisión clínica, pudiendo desencadenarse exacerbaciones por factores como la exposición solar, infecciones, intervenciones quirúrgicas,…Las esclerodermias localizadas suponen una excepción ya que la duración media de la actividad suele estar limitada a 3-5 años y la progresión a esclerosis sistémica es excepcional. El pronóstico va a estar determinado por los órganos afectados y la toxicidad de los tratamientos recibidos. El LES en niños y adolescentes tiene peor pronóstico que los casos de inicio en la edad adulta, ya que presentan manifestaciones clínicas más graves y mayor morbilidad (osteoporosis, aterosclerosis y enfermedad coronaria precoces) y mortalidad (5-15% de los casos), principalmente por complicaciones infecciosas o fracaso renal. La mayor parte de los adolescentes con dermatomiositis juvenil realizará una vida normal, aunque no todos podrán realizar una actividad física intensa, siendo la mortalidad baja (1-2% casos), normalmente debido a complicaciones respiratorias.
Tablas y Figuras
Tabla 1. Clasificación de la nefritis lúpica
|
Nefritis lúpica |
Descripción |
Histología |
|
Clase I |
GN mesangial con cambios mínimos |
Glomérulos normales al MO pero con depósitos inmunes en la IF |
|
Clase II |
GN mesangial proliferativa |
Hipercelularidad mesangial con expansión de la matriz mesangial y depósitos inmunes en el mesangio |
|
Clase III |
GN focal |
GN focal, segmentaria o global que afecta a menos del 50% de los glomérulos con depósitos inmunes subendoteliales difusos |
|
Clase IV |
GN difusa |
GN difusa, segmentaria o global que afecta a más del 50% de los glomérulos con depósitos inmunes subendoteliales difusos |
|
Clase V |
GN membranosa |
GN global o segmentaria con depósitos inmunes subepiteliales |
|
Clase VI |
GN esclerosante avanzada |
≥90% de los glomérulos están esclerosados y sin actividad residual |
GN: glomerulonefritis; MO: microscopía óptica; IF: inmunofluorescencia
Tabla 2. Criterios diagnósticos de lupus eritematoso sistémico
|
Criterio |
Definición |
|
1. Eritema malar |
Eritema fijo, liso o elevado, en “alas de mariposa”, con tendencia a respetar surco nasogeniano |
|
2. Eritema discoide |
Placas eritematosas elevadas con hiperqueratosis; puede existir cicatrización atrófica en lesiones antiguas |
|
3. Fotosensibilidad |
Exantema tras la exposición solar, recogido en la historia o documentado por un médico |
|
4. Úlceras orales |
Ulceraciones orales o nasofaríngeas, no dolorosas |
|
5. Artritis |
Artritis no erosiva de articulaciones periféricas |
|
6. Serositis |
Pleuritis o pericarditis |
|
7. Trastornos renales |
Proteinuria persistente >0.5 g/dL o Cilindros celulares |
|
8. Trastornos neurológicos |
Convulsiones o psicosis en ausencia de causa metabólica o medicamentosa |
|
9. Trastornos hematológicos |
Anemia hemolítica con reticulocitosis o Leucopenia < 4000/μL en 2 o más ocasiones o Linfopenia < 1500/μL en 2 o más ocasiones o Trombocitopenia < 100.000/μL |
|
10. Trastornos inmunológicos |
Anticuerpos antiADN o Anticuerpos antiSm o Anticuerpos antifosfolípido (anticuerpos anticardiolipina, presencia de anticoagulante lúdico o VDRL falso +) |
|
11. Anticuerpos antinucleares |
Por inmunofluorescencia o técnica equivalente |
Tabla 3. Criterios para el diagnóstico de dermatomiositis juvenil
|
Criterios: |
|
1. Debilidad simétrica de la musculatura proximal (cinturas, flexora cervical) 2. Biopsia muscular con evidencia de necrosis, fagocitosis, regeneración, atrofia perifascicular, variación en el tamaño de las fibras musculares, infiltrado inflamatorio perivascular 3. Elevación sérica de enzimas musculares 4. Alteraciones electromiográficas demostrando evidencia de miopatía y denervación (potenciales de unidad motora polifásicos de baja amplitud y corta duración;fibrilaciones;descargas repetitivas de alta frecuencia) 5. Manifestaciones cutáneas características (rash heliotropo, pápulas/signo de Gottron) |
|
Diagnóstico: |
|
DMJ “definida”: cambios cutáneos + 3 criterios adicionales DMJ “probable”: cambios cutáneos + 2 criterios adicionales DMJ “posible”: cambios cutáneos + 1 criterio adicional |
Tabla 4. Criterios para la clasificación de la esclerosis sistémica juvenil
|
Criterio mayor (requerido) |
|
Induración/engrosamiento de la piel proximal a las articulaciones metacarpofalángicas o metatarsofalángicas |
|
Criterios menores (2 requeridos) |
|
Cutáneos: esclerodactilia Vasculares periféricos: fenómeno de Raynaud, alteraciones del lecho ungueal (megacapilares o áreas avasculares) o úlceras en la punta de los dedos Gastrointestinales: disfagia o reflujo gastroesofágico Cardiacos: arritmias, fallo cardiaco Renales: crisis renal esclerodérmica, hipertensión arterial de reciente comienzo Respiratorios: fibrosis pulmonar (en radiografía de tórax o en TC alta resolución), DLCO disminuido, hipertensión arterial pulmonar (primaria o secundaria a enfermedad pulmonar intersticial, valorada por ecocardiograma) Neurológicos: neuropatía o síndrome del túnel del carpo Musculoesqueléticos: artritis, miositis o roce en tendones Serológicos: anticuerpos antinucleares o autoanticuerpos selectivos de esclerosis sistémica (antitopoisomerasa 1 o Scl-70, anticentromero, anti ARN polimerasa I o III, anti PM-Scl, antifibrilina) |
Sensibilidad del 90% y especificidad del 96% cuando el criterio mayor y 2 criterios menores están presentes; TC: tomografía computerizada; DLCO: capacidad de difusión del monóxido de carbono
Tabla 5. Fármacos utilizados en el tratamiento del lupus eritematoso sistémico
|
FÁRMACO |
INDICACIONES |
Dosis |
Observaciones |
|
AINEs |
Manifestaciones musculoesqueléticas Síntomas constitucionales leves Pleuritis o pericarditis leve |
Variable según AINE utilizado |
Meningitis aséptica |
|
Hidroxicloroquina |
Manifestaciones cutáneas, alopecia Artritis, enfermedad sistémica leve Terapia adyuvante en la mayoría de pacientes |
5-6 mg/kg/día (máx 400 mg/día) |
Requiere controles oftalmológicos anuales |
|
Glucocorticoides |
En la mayoría de pacientes |
Variable |
|
|
Metotrexato |
Manifestaciones musculoesqueléticas o cutáneas Agente ahorrador de esteroides |
10-15 mg/m2/sem VO o vía SC |
Suplementar con ácido folínico |
|
Azatioprina |
Nefritis lúpica clase III o IV Manifestaciones neuropsiquiátricas Agente ahorrador de esteroides |
3 mg/kg/día (máx 150 mg/día) |
Considerar determinación de actividad o polimorfismos de tiopurinametiltransferasa |
|
Micofenolato mofetilo (MMF) o Ácido micofenólico (AMF) |
Nefritis lúpica clase III o IV Manifestaciones neuropsiquiátricas Agente ahorrador de esteroides (si fallo o intolerancia a metotrexato o azatioprina) |
MMF: 1 g/m2/día en 2 dosis VO AMF: 720 mg/m2/día en 2 dosis VO |
Ajustar dosis según tolerancia Monitorizar niveles |
|
Ciclofosfamida |
Manifestaciones neuropsiquiátricas Nefritis lúpica clase III o IV |
500-1000 mg/m2 IV |
Hidratación previa a administración |
|
Ciclosporina |
Nefritis lúpica clase V Síndrome de activación del macrófago |
2-5 mg/kg/día en 2 dosis VO |
Monitorizar niveles |
AINEs: antiinflamatorios no esteroideos; VO: vía oral; SC: subcutánea; IV: intravenosa
Figura 1. Exantema malar (lupus eritematoso sistémico)
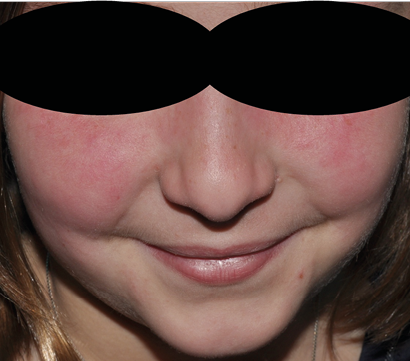
Figura 2. Pápulas de Gottron (dermatomiositis juvenil)
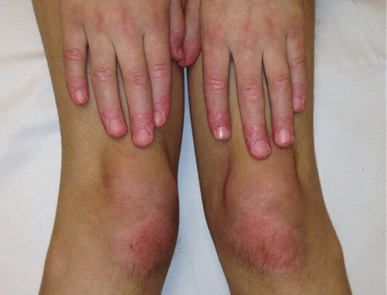
Figura 3. Eritema heliotropo (dermatomiositis juvenil)
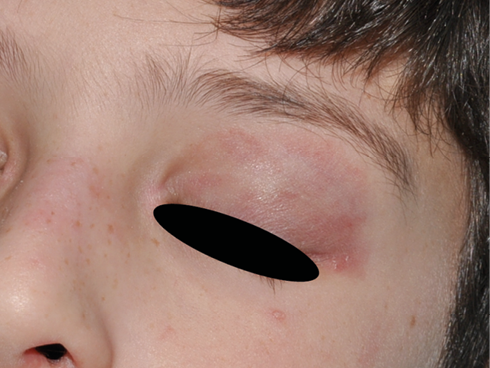
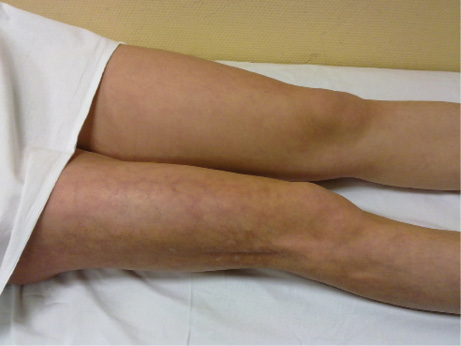
Figura 4. Esclerodermia lineal
Bibliografía
1. Tucker LB. The young person with systemic lupus erythematosus. En McDonagh JE, White PH ed. Adolescent rheumatology. Informa health care, New York 2008: p169-181.
2. Kumar TS, Aggarwal A. Approach to a patient with connective tissue disease. Indian J Pediatr 2010; 77:1157-1164.
3. Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 1997;40(9):1725.
4. Weening JJ, D´Agati VD, Scwartz MM, et al. The classification of glomerulonephritis in systemic lupus erythematosus revisited. J Am Soc Nephrol 2004;15:241-50.
5. Levy DM, Kamphuis S. Systemic Lupus erythematosus in children and adolescents. Pediatr Clin N Am 2012; 59: 345-64.
6. Weiss JE. Pediatric systemic lupus erythematosus: more than a positive antinuclear antibody. Pediatr Rev 2012;33(2):62-73.
7. Silverman E, Eddy A. Systemic lupus erythematosus. En Cassidy JT, ed.Textbook of pediatric rheumatology. Saunders Elsevier, Philadelphia 2011:p315-343.
8. McCann LJ, Sen D. The disease spectrum of adolescent rheumatology. En McDonagh JE, White PH ed. Adolescent rheumatology. Informa health care, New York 2008: p107-147.
9. Bohan A, Peter JB. Polymiositis and dermatomyositis (first of two parts). N Engl J Med 1975; 292:344-7.
10. Huber AM. Idiopathic inflammatory myopathies in childhood: current concepts. Pediatr Clin N Am 2012; 59: 365-80.
11. Rider LG, Lindsley CB, Cassidy JT. Juvenile dermatomiositis. En Cassidy JT, ed.Textbook of pediatric rheumatology. Saunders Elsevier, Philadelphia 2011:p375-413.
12. Laxer RM, Zulian F. Localized sclerodermas. Curr Opin Rheumatol 2006;18(6):606-13.
13. Zulian F, Woo P, Athreya BH et al. The Pediatric Rheumatology European Society/America College of Rheumatology/European League against Rheumatism provisional classification criteria for juvenile systemic sclerosis. Arthritis rheum 2007;57(2):203-12.
14. Torok KS. Pediatric scleroderma: systemic or localized forms. Pediatr Clin N Am 2012; 59: 381-45.
15. Zulian F, Cassidy JT. The systemic sclerodermas and related disorders. En Cassidy JT, ed.Textbook of pediatric rheumatology. Saunders Elsevier, Philadelphia 2011:p414-437.
16. Pepmueller PH, Linsley CB, Cassidy JT. Mixed connective tissue disease and undifferentiated connective tissue disease. En Cassidy JT, ed.Textbook of pediatric rheumatology. Saunders Elsevier, Philadelphia 2011:p448-457.

