Enfermedades renales más habituales en la adolescencia
V.M. García Nieto, T. Moraleda Mesa, P. Tejera Carreño, M.I. Luis Yanes.
Sección de Nefrología Pediátrica, Hospital Universitario Nuestra Señora de Candelaria, Santa Cruz de Tenerife.
Fecha de recepción: 29 de septiembre 2019
Fecha de publicación: 31 de octubre 2019
Adolescere 2019; VII (3): 6-21
Resumen
|
Algunas enfermedades renales pueden debutar en la adolescencia. Tal es el caso de la enfermedad de Gitelman, tubulopatía que cursa con hipomagnesemia e hipocalciuria. Las glomerulopatías que con más frecuencia requieren una biopsia renal en esa edad son las nefropatías lúpica e IgA y el síndrome nefrótico resistente a corticoides. En adolescentes mujeres pueden observarse casos de infecciones urinarias originadas por Staphylococcus saprophyticus. No deben olvidarse, asimismo, las infecciones de transmisión sexual. Respecto a las anomalías metabólicas causantes de cálculos, con cierta frecuencia los pacientes con hipercalciuria idiopática normalizan en la adolescencia la eliminación urinaria de calcio pero reducen, también, la de citrato. Los casos con diabetes tipo 1 que tienen un debut muy precoz, pueden presentar en esa edad los primeros datos de nefropatía, especialmente, si el control metabólico ha sido inadecuado durante la infancia. La diabetes tipo 2 puede debutar en la adolescencia asociada, en general, a obesidad.
La monitorización ambulatoria de la presión arterial es útil porque repara varias de las limitaciones de las medidas casuales de la presión arterial en la consulta. Algunos pacientes portadores de malformaciones renales que cursan con pérdida de parénquima, pueden mostrar un empeoramiento del filtrado glomerular renal en la adolescencia.
Palabras clave: Adolescencia; Enfermedad de Gitelman; Diabetes; Obesidad; Hipertensión Arterial.
|
Abstract
|
Certain kidney diseases can present in adolescence. Such is the case of Gitelman disease, a tubulopathy with hypomagnesemia and hypocalciuria. The glomerulopathies that most frequently require a renal biopsy during that age are lupus and IgA nephropathies as well as steroid-resistant nephrotic syndrome. In female adolescents, urinary infection caused by Staphylococcus saprophyticus can be observed. Sexually transmitted infections should not be forgotten. Among the metabolic abnormalities that cause renal stones, patients with idiopathic hypercalciuria tend to normalize urinary calcium elimination during adolescence but also reduce that of citrate. Patients with type 1 diabetes with a very early onset, especially if the metabolic control has been inadequate during childhood, may manifest the first data of nephropathy during adolescence. Type 2 diabetes mellitus may present in adolescence, generally associated with obesity. Ambulatory blood pressure monitoring is useful because it addresses several of the limitations of casual blood pressure measurements in the office. Some patients with renal malformations who suffer parenchymal loss may show a worsening of glomerular filtration rate in adolescence.
Key words: Adolescence; Gitelman disease; Diabetes; Obesity; Hypertension.
|
Introducción
Escribir un texto sobre las nefropatías más frecuentes en la adolescencia es un reto, porque existen pocas publicaciones relacionadas con algo tan específico. Para seleccionar los temas a tratar hemos tenido que recurrir, básicamente, a las particularidades que hemos observado en nuestra práctica diaria. Así, hemos seleccionado entidades que debutan, en general, en la adolescencia como la enfermedad de Gitelman o algunas glomerulopatías. De algunos temas generales hemos intentado destacar sus características en la adolescencia como es el caso de las infecciones de vías urinarias y la urolitiasis. Otras enfermedades, en fin, empiezan a desplegar sus complicaciones en la adolescencia. Es el caso de la nefropatía diabética, la hipertensión arterial ligada, sobre todo, a la obesidad y las malformaciones que cursan con pérdida de parénquima renal.
Infección de vías urinarias
Definición. Factores de riesgo
La infección de vías urinarias (IVU) es la entidad clínica que se caracteriza por el crecimiento de microorganismos (generalmente bacterias) en las vías urinarias con síntomas compatibles.
La IVU se produce fundamentalmente por vía ascendente, como resultado de la colonización del área periuretral por microorganismos que suelen provenir de la flora fecal.
Se han descrito distintos factores que aumentan el riesgo de IVU. Entre ellos destacan:
Anomalías que ocasionan obstrucción del flujo urinario (estenosis pieloureteral, megauréter, vejiga neurógena…)
- Instrumentación de la vía urinaria
- Diabetes mellitus mal controlada
- Predisposición genética
- Varones no circuncidados
- Uso de espermicidas y/o diafragma como método anticonceptivo
- Actividad sexual
Por otra parte, se han identificado algunos factores que en los casos de IVU, incrementan la posibilidad de formar cicatrices renales secundarias. Dichos factores son la fiebre elevada (>39ºC), la presencia de alteraciones ecográficas, el retraso en la instauración del tratamiento antibiótico más allá de las primeras 72 horas tras el inicio de los síntomas y la etiología distinta a Escherichia coli(1).
Etiología
La bacteria que causa IVU con mayor frecuencia (75-95% de los casos) es Escherichia coli (Tabla I).
Diagnóstico
La orientación diagnóstica inicial se basa fundamentalmente en la clínica y en la exploración física del paciente. Si la clínica es compatible con una infección urinaria se recogerá una muestra de orina limpia de mitad del chorro miccional para realizar un análisis de orina en todos los casos y urocultivo.
En cuanto a la forma de recogida de la muestra, la técnica debe realizarse tras retracción prepucial o separación de los labios mayores y limpieza con suero o jabón suave de la zona, desechando la primera y última parte de la micción.
El resultado del análisis urinario y la sintomatología pueden ayudar a decidir si es necesario iniciar tratamiento antibiótico de manera empírica sin esperar el resultado del cultivo de orina (48 horas) (Tabla II).
El cultivo de orina no será necesario en mujeres sin patología de base en las que se sospeche una cistitis; en este caso, puede ser suficiente para iniciar el tratamiento antibiótico la presencia de leucocituria junto a síntomas clínicos compatibles. Debe recogerse urocultivo en todos los casos en los varones y en aquellos pacientes que presenten patología nefrourológica de base, independientemente del sexo, así como en personas con IVU recurrentes o en las que sospechemos por la clínica una pielonefritis.
El cultivo de orina se considera positivo cuando existan ≥ 100.000 UFC/campo.
Peculiaridades en la adolescencia
La adolescencia es una época de cambios constantes en cuya etapa se suelen iniciar las relaciones sexuales. Por este motivo, los pediatras no deben olvidarse de pensar en las enfermedades de transmisión sexual como diagnóstico diferencial de IVU en aquellos adolescentes sexualmente activos,
siendo imprescindible una anamnesis detallada y una exploración genital en busca de úlceras, secreción uretral o secreción cervical. En las adolescentes mujeres debemos también considerar la vulvovaginitis como parte del diagnóstico diferencial, pues pueden presentar una clínica similar a una IVU.
El aislamiento de algunos microorganismos en mujeres adolescentes como estafilococos coagulasa negativos distintos de Staphylococcus saprophyticus(2), estreptococos del grupo B y lactobacilos representa con mucha frecuencia la contaminación de la muestra de orina.
Particularmente, Staphylococcus saprophyticus es resistente a los medicamentos más utilizados para el tratamiento empírico de las IVU. En una revisión reciente, se observó que muchas IVU fueron tratadas con un antibiótico empírico que no fue efectivo contra esa bacteria, revelando que S. saprophyticus es una etiología que no se considera suficientemente(2). Afortunadamente, la fosfomicina, de uso habitual en nuestro país, tiene una buena actividad in vitro contra Staphylococcus saprophyticus y otros uropatógenos comunes, como Escherichia coli, Proteus mirabilis y Klebsiella pneumoniae(3). El 94% de las bacterias de nuestro hospital causantes de IVU en los años 2013 y 2014 fueron sensibles a fosfomicina (Can Pediatr 2016; 40:19-22).
La fosfomicina, de uso habitual en nuestro país, tiene una buena actividad in vitro contra Staphylococcus saprophyticus y otros uropatógenos comunes
Tratamiento de las infecciones urinarias en la adolescencia(4)
Cistitis
De elección: fosfomicina trometamol 3 g en dosis única. Puede repetirse a las 48 o 72 horas si persisten los síntomas.
Alternativas:
- Nitrofurantoína 100 mg/12 h durante 5 días.
- Cotrimoxazol 800/160 mg /12 h durante 3 días.
- Ciprofloxacino 250 mg/12 h durante 3 días.
Pielonefritis
De elección: Cefixima 400mg /24 h durante 7 días.
Alternativas:
- Ciprofloxacino 750 mg/12 h durante 7 días.
- Amoxicilina/clavulánico 875/125mg cada 8 horas durante 7 días.
Resistencias bacterianas
Las tasas de resistencia de E. coli han aumentado en todo el mundo, aunque varían mucho según la zona geográfica. Las tasas de resistencia para ampicilina en la mayoría de las zonas estudiadas son superiores al 20% y en otras muchas, también, para la trimetoprim (con o sin sulfametoxazol), por lo que deben evitarse como tratamiento inicial. La nitrofurantoína y la fosfomicina han demostrado una buena actividad in vitro en todos los países investigados. Las tasas de resistencia de las cefalosporinas orales de primera y segunda generación y de la amoxicilina-ácido clavulánico son variables regionalmente pero, generalmente, menores al 10%, por lo que son una buena opción de tratamiento.
Las tasas de resistencia de las fluoroquinolonas están también por debajo del 10% en Europa y América del Norte, aunque se ha observado una clara tendencia a su incremento en los últimos años.
La monitorización continua de las tasas de resistencia a nivel local es fundamental para poder optimizar el tratamiento empírico.
Litiasis renal
Introducción
La litiasis renal es una de las primeras enfermedades claramente identificadas en el ser humano.
Un estudio reciente, evidencia una prevalencia global de litiasis renal del 15,5% en población española de 40 a 65 años(5). Otros estudios demuestran que su prevalencia está en un claro incremento.
La composición de los cálculos, su ubicación en el tracto urinario y la prevalencia de la enfermedad varía en todo el mundo. La tasa de prevalencia en países de ingresos bajos a medios como Pakistán y Turquía es del 5% al 15%, en comparación con el 1% al 5% en los países de superior nivel económico. Los cálculos urinarios pueden ser localizados en cualquier parte del tracto urinario. Muchos cálculos encontrados en niños nacidos en países de ingresos bajos a medios se encuentran dentro de la vejiga urinaria.
La manifestación y la presentación clínica de cálculos urinarios en niños difieren de la población adulta y puede variar con la edad. El 50% de los niños presentarán dolor abdominal, el 33% con hematuria y el 11% con infección. El dolor en niños con cálculos puede tener una distribución como apendicitis o gastroenteritis. En la práctica clínica, los ultrasonidos tienen una alta especificidad para detectar la nefrolitiasis en niños, pero con una sensibilidad moderada (Figura 1).
Fisiopatología
La litiasis renal puede definirse como una alteración de las condiciones naturales de cristalización de la orina. El tiempo que se requiere para generar un cristal depende fundamentalmente de la sobresaturación de la disolución (exceso de soluto en la disolución: fuerza impulsora de la cristalización), de la presencia de partículas sólidas preexistentes (los llamados nucleantes heterogéneos) y de la presencia de inhibidores de la cristalización. Estos últimos son sustancias que debido a su estructura química interaccionan con el núcleo o las caras del cristal, interfiriendo notablemente en su formación o/y desarrollo, reduciendo o previniendo los procesos de cristalización. Todas las orinas humanas están sobresaturadas con respecto al oxalato cálcico(6), de tal manera que el grado de sobresaturación resulta más elevado en los individuos hipercalciúricos o/y hiperoxalúricos. La orina humana puede contener además una amplia variedad de nucleantes heterogéneos tales como agregados proteicos, residuos celulares o bacterias. Además, en este aspecto, también debe considerarse la capacidad nucleante de los epitelios renales alterados. Es evidente que al aumentar el tiempo de permanencia de la orina en el tracto urinario (principalmente en las vías altas), se incrementa la posibilidad de que los procesos de cristalización conduzcan a la formación de cálculos renales. La existencia de cavidades renales de baja eficacia urodinámica constituye un importante factor de riesgo del desarrollo de cálculos. De hecho, se ha demostrado que los factores morfoanatómicos pueden jugar un importante papel en la calculogénesis. Así, se explicaría que un paciente recidivante en el que es de suponer que la orina tendrá la misma composición en los dos riñones, sólo forme cálculos en uno de ellos(7). Cuando el desarrollo de cristales se produce en la vejiga urinaria, normalmente se eliminan sin dificultad como cristaluria asintomática.
Clasificación de los cálculos renales
Independientemente de su composición química, los cálculos renales pueden clasificarse de manera amplia en dos grandes categorías: cálculos formados sobre las paredes renales (unidos a las papilas) en los que claramente se distingue la zona de unión al epitelio y cálculos desarrollados en las cavidades renales (sin zona de unión al epitelio)(6). Los cálculos más frecuentes son los de oxalato cálcico, fosfato (Figura 2), urato y cistina renales úricos. Existen otros cálculos poco frecuentes (Figura 3), la mayoría de los cuales están en relación con fármacos poco solubles como triamtereno, indinavir, sílice, glefamina o sulfamidas.
La infección bacteriana prolongada del tracto urinario suele ser la causa más común de este tipo de litiasis. Los gérmenes ureolíticos (Proteus, Klebsiella, Pseudomona, Ureaplasma…) suelen provocar una notable elevación del pH urinario (pH> 7) y de la concentración urinaria de amonio, que conjuntamente favorecen la precipitación de fosfato amónico magnésico y de hidroxiapatita (fosfato de calcio). Detalle de un cristal de estruvita en cuyas caras se observan las marcas en “Y” que permiten su rápida identificación junto pequeñas zonas de esferulitos de hidroxiapatita (cortesía del Dr. Félix Grases. Laboratorio de Investigación en Litiasis Renal, Instituto Universitario de Investigación en Ciencias de la Salud, Universidad de las Islas Baleares, Palma de Mallorca, España).
Paciente que a los 14 años expulsó tres cálculos. En el estudio metabólico se observó hipocitraturia. Vista general y detalle al microscopio electrónico de barrido del interior del “cálculo” de carbonato cálcico. La estructura no se corresponde con la típica de un cálculo renal con una morfología correspondiente a una estructura de origen vegetal, por lo que podría tratarse de una semilla (Dr. Grases).
Anomalías metabólicas causantes de cálculos
En la práctica clínica suelen determinarse calcio, ácido úrico, oxalato, cistina, citrato y magnesio.
Los cuatro primeros favorecen la formación de cálculos cuando su concentración es elevada en la orina (favorecedores). En cambio, citrato y magnesio propician su formación cuando sus cantidades urinarias son reducidas (inhibidores). Su cuantificación se realiza en orina de 24 horas. Al menos, en pediatría, se debe confirmar que la recogida horaria urinaria es correcta mediante el cálculo de la eliminación urinaria de creatinina (normal: 15-25 mg/kg/día). No obstante, en la actualidad, cada vez se usan más los cocientes urinarios por su facilidad en la recogida de las muestras, especialmente, en la infancia y porque la concentración, especialmente, de calcio y citrato pueden variar en distintos momentos del día. En todo caso, los cocientes urinarios son muy útiles, especialmente, en el seguimiento de los pacientes. En el diagnóstico y control de los pacientes litiásicos es muy útil el cociente calcio/citrato. La orina es particularmente litógena cuando existe un desequilibrio entre el componente favorecedor (calcio) y el protector (citrato). Valores de ese cociente superiores a 0,33 indican que la orina es potencialmente litógena, independientemente de la edad y del momento de la recogida. Nosotros, hemos observado que los cocientes urinarios calcio/creatinina, citrato/creatinina y calcio/citrato son distintos en dos momentos del día (orina de la noche recogida antes de la cena y primera de la mañana siguiente), de tal modo que las orinas más litógenas son las que se forman por la noche, es decir, las recogidas por la mañana (primera orina del día)(7). La consecuencia de todo ello es que estos datos tan sensibles no se pueden reproducir en la orina recogida durante 24 horas.
- Hipercalciuria
La causa más habitual de litiasis tanto en niños como en adultos es la hipercalciuria idiopática (HI). Se define por la ausencia de hipercalcemia y de otras causas identificables de hipercalciuria secundaria. La HI es de origen genético. No se considera una enfermedad en si misma sino una anomalía metabólica puesto que, en muchas ocasiones, no se asocia con clínica ni con formación de cálculos. El mecanismo fisiopatológico de la HI es muy complejo. La hipótesis más aceptada en la actualidad acerca de la causa de la HI está en relación con la existencia de un incremento del número de receptores para la vitamina D tanto en las células intestinales y óseas (osteoclastos) como en los monocitos periféricos.
- Hiperoxaluria. Oxalosis
- Hiperuricosuria
- Cistinuria
- Hipocitraturia
El citrato actúa como un inhibidor de la formación de cálculos de calcio al formar un complejo soluble, lo que disminuye la disponibilidad del calcio iónico libre, necesario para la cristalización de oxalato o de fosfato cálcicos. El citrato, también actúa como un inhibidor directo de la agregación de cristales de calcio y de su crecimiento. Por tanto, un citrato urinario reducido puede ser una causa importante de litiasis cálcica. En general, la acidosis metabólica se acompaña de hipocitraturia y la alcalosis de hipercitraturia. Por ello, las principales causas de eliminación urinaria reducida de citrato son la acidosis tubular renal y la insuficiencia renal crónica. Con cierta frecuencia, se observa hipocitraturia asociada a hipercalciuria idiopática, en ausencia de acidosis tubular renal. La ingesta excesiva de proteínas también favorece su aparición por la sobrecarga ácida que ocasiona.
- Hipomagnesuria
- Otros inhibidores de la formación de cálculos
Además del citrato y el magnesio, otras sustancias como pirofosfato, ciertos glicosaminoglicanos, nefrocalcina y fitato actúan inhibiendo la formación de cristales de oxalato de calcio y de fosfato de calcio. Por tanto, cuando sus niveles son bajos, se favorece la formación de cristales. El fitato, presente en la cáscara de los cereales y en legumbres, es un potente inhibidor de la cristalización. En este sentido, se recomienda el consumo frecuente de cereales integrales en pacientes con litiasis de repetición.
Prelitiasis
Desde principios de los 80, los pediatras hemos aprendido a identificar a los niños portadores de anomalías metabólicas causantes de cálculos en un momento en el que aún no les ha dado tiempo a fomarlos. Esto es particularmente cierto con las dos causas más frecuentes de los mismos, la hipercalciuria idiopática y la hipocitraturia. La hipercalciuria es de origen genético. Por tanto, uno de los dos padres es portador de la misma anomalía haya tenido o no síntomas de enfermedad litiásica. Los niños con HI pueden debutar con síntomas o signos como hematuria macro o microscópica, disuria estéril, polaquiuria, urgencia miccional, incontinencia urinaria, enuresis nocturna, orinas turbias, dolor abdominal recurrente “no típico de cólico renal” o leucocituria estéril(8). La hipocitraturia es una cuestión pendiente pues, si bien, en ocasiones se reconoce su causa en otras se desconoce y puede coincidir o no con la hipercalciuria.
Particularidades en la adolescencia
Los niños con prelitiasis pueden formar microcálculos que son difíciles de observar en la ecografía. Pueden crecer y aparecer en la vía urinaria produciendo un cólico nefrítico. Eso ocurre particularmente si en determinado momento no se toman las medidas dietéticas preventivas que figuran más abajo.
En nuestra práctica diaria hemos observado que este hecho es relativamente frecuente en adolescentes.
Con mucha asiduidad, se observa que los niños con hipercalciuria idiopática, al acercarse a la adolescencia, normalizan la eliminación urinaria de calcio y muestran, entonces, hipocitraturia. Parece, pues, por tanto, como si ambas anomalías metabólicas tuvieran un origen común. Una situación particular puede ocurrir, preferentemente, en la adolescencia cuando la citraturia puede estar reducida y la calciuria también, con lo que la orina no es litogena al mostrar una relación calcio/citrato normal.
Por esta razón, algunos adolescentes con prelitiasis que no se cuidan adecuadamente, pueden no formar cálculos durante esos años de crecimiento intenso y formación incrementada de hueso debido a una mayor actividad osteoblástica.
Con mucha asiduidad, se observa que los niños con hipercalciuria idiopática, al acercarse a la adolescencia, normalizan la eliminación urinaria de calcio y muestran, entonces, hipocitraturia
Algunos pacientes con enfermedades crónicas que cursan con predisposición a formar cálculos renales pueden manifestarse en la adolescencia con hematuria macroscópica o cólicos nefríticos. Son paradigmáticos los casos de la fibrosis quística y de la enfermedad inflamatoria intestinal en los que es frecuente la presencia de hipocitraturia.
Se han descrito casos de litiasis que han ofrecido los primeros síntomas en la adolescencia como expresión de riñón en esponja (enfermedad de Cacchi Ricci), riñón en herradura o hiperparatiroidismo primario. También, se ha descrito una asociación en esa edad entre nefrolitiasis e incremento del índice de masa corporal. Así, los adolescentes obesos tienen una mayor probabilidad de formar cálculos renales asociado, asimismo, con hipocitraturia.
Los adolescentes obesos tienen una mayor probabilidad de formar cálculos renales asociado a hipocitraturia
Tratamiento dietético preventivo
La mayoría de las anomalías metabólicas causantes de cálculos tienen un origen genético, por lo que la predisposición litiásica dura toda la vida. Por tanto, debe intentarse un control dietético y reservarse el tratamiento farmacológico para los casos complicados. El tratamiento dietético, a nivel general, incluye una ingesta elevada de agua (2000-3000 ml/1.73 m2), de frutas (cítricos) y de verduras, pescado azul y cereales integrales. A la inversa, no se debe abusar de proteínas ni de sal.
La citraturia se eleva incrementando la ingesta de agua y de cítricos y reduciendo la de proteínas de origen animal y de sal.
Nefropatía diabética
Generalidades
Antes del descubrimiento de la insulina por Banting y Best en 1921, las complicaciones crónicas de la diabetes mellitus (DM) no eran tan frecuentes como en la actualidad ya que los pacientes diabéticos fallecían antes de que estos problemas se hicieran manifiestos. Actualmente se sabe que no hay órgano o sistema que escape de estar involucrado por la DM.
Muchas investigaciones han demostrado que en la niñez se pueden observar alteraciones que expresan daño microvascular y, aunque la mayoría de estas no se manifiestan clínicamente en la edad pediátrica, en esta época de la vida se pueden poner en marcha los mecanismos fisiopatológicos que acabarán manifestándose clínicamente en la edad adulta. Una de estas complicaciones es la nefropatía diabética, que fue reconocida en 1936 cuando Kimmestiel y Wilson, dos anatomopatólogos alemanes, descubrieron la forma nodular de esta glomerulopatía.
El síndrome clínico resultante de todas estas alteraciones se caracteriza por proteinuria persistente, hipertensión arterial y deterioro progresivo de la función renal.
La variedad preferente de afectación en la población pediátrica es la DM insulindependiente o tipo 1 (DM-1). Sin embargo, es necesario resaltar que en las últimas décadas se ha observado un importante incremento en la prevalencia de DM tipo 2 (DM-2) en niños y adolescentes. La creciente tasa de obesidad es el factor principal que explica esta situación. Así, se objetiva obesidad o sobrepeso en más del 95% de los adolescentes con DM-2. Además, existe historia familiar de DM-2 en el 90% de los casos, siendo sintomáticos en el momento de su presentación más de las dos terceras partes de los adolescentes, con más de un 13% en los que se objetiva la presencia de albuminuria en el momento del diagnóstico (el término “microalbumina” es incorrecto puesto que no se trata de una molécula más pequeña sino de cantidades reducidas). Este problema parece ir en aumento, y se estima que en el momento actual, entre un 20-30% de los nuevos casos de DM en la edad pediátrica corresponden a DM-2.
Es necesario resaltar que en las últimas décadas se ha observado un importante incremento en la prevalencia de DM tipo 2 (DM-2) en niños y adolescentes. La creciente tasa de obesidad es el factor principal que explica esta situación
La nefropatía diabética es un determinante de primer orden del exceso de morbilidad y de mortalidad prematura asociada a la DM-1. En el momento actual, con el control intensivo desde el punto de vista metabólico y de la presión arterial, y con el uso de estrategias farmacológicas con demostrada capacidad renoprotectora, las tasas de nefropatía diabética establecida y enfermedad renal terminal han descendido respecto a etapas anteriores(9). La incidencia acumulativa de nefropatía diabética aumenta progresivamente hasta aproximadamente los 20 años de evolución de la diabetes, cuando alcanza su punto máximo, para descender a partir de este momento, sugiriendo que el riesgo de desarrollar esta complicación no es constante a lo largo del curso de la enfermedad.
Detección precoz de la nefropatía diabética incipiente
Es fácilmente entendible la importancia de contar con un método simple y reproducible que permita discriminar a los pacientes con DM-1 en función del riesgo de desarrollar nefropatía. Desde esta perspectiva, se ha demostrado la utilidad del estudio del ritmo circadiano de la presión arterial. Así, una alteración de este patrón circadiano, en concreto, una elevación en la presión arterial nocturna, precede a la aparición de albuminuria. Se ha descrito que un descenso normal nocturno de la presión arterial tiene un valor predictivo negativo del 91% para el desarrollo de albuminuria, con una reducción del 70% en el riesgo de presentar esta complicación(10).
En el momento actual, el marcador aceptado como capaz de identificar precozmente a los pacientes que asocian un alto riesgo para desarrollar nefropatía diabética es la albuminuria persistente. Para un diagnóstico correcto, la albuminuria ha de ser positiva en dos de tres determinaciones consecutivas realizadas en un período de tres a seis meses. La cuantificación de la excreción de albúmina en orina de 24 horas es considerada el patrón de oro, aunque también podrá realizarse la determinación en orina minutada o en una muestra de orina aislada. Este último método, donde se determina el índice albúmina/creatinina (normal: menor de 30 mg/g), ha mostrado resultados superponibles a los obtenidos en orina de 24 horas, evitando el posible error derivado de una recolección incompleta de orina. El uso de este cociente ha sido recomendado como la estrategia de despistaje en todos los pacientes diabéticos.
Características en la adolescencia
Cuando la diabetes se inicia precozmente, al llegar a la adolescencia pueden observarse los primeros datos de nefropatía, especialmente, si el control de la enfermedad ha sido inadecuado durante la infancia.
Si se diagnostica diabetes en niños pequeños y la duración prepuberal de la diabetes es muy larga, los pacientes parecen estar protegidos contra la retinopatía diabética. Esta protección desaparece si el control metabólico ulterior es malo. En cambio, cuando el inicio es en la pubertad, el riesgo de retinopatía diabética es mayor y menos dependiente del control metabólico y puede estar influenciado por factores relacionados con la edad, como la presión arterial.
La ocurrencia de eventos importantes en la vida de los pacientes se asoció con una peor atención de la diabetes y con más cualidades psicosociales negativas en adolescentes con diabetes tipo 1(11). Como se ha indicado más arriba, en las últimas décadas se ha observado un importante incremento en la prevalencia de DM tipo 2.
Consideraciones terapéuticas
Desde el punto de vista terapéutico, la presencia de albuminuria representa un estadio evolutivo de la nefropatía diabética en la cual, el tratamiento es efectivo para prevenir la progresión de la enfermedad renal. Una duración corta de la albuminuria, unas bajas concentraciones séricas de colesterol y triglicéridos y un adecuado control metabólico y de la presión arterial, principalmente de la presión arterial sistólica, se han identificado como factores que se asocian de forma independiente con la regresión de la albuminuria(12). El tratamiento farmacológico inicial de elección será un inhibidor del enzima de conversión de la angiotensina (IECA), con una dosis que podrá ser progresiva hasta alcanzar la dosis máxima efectiva recomendada. Es de interés destacar que este tratamiento ha de establecerse en todo paciente con albuminuria, tanto hipertensos como normotensos, dado que se consigue una reducción precoz de la hiperfiltración glomerular, de la presión intraglomerular, de la albuminuria y de la progresión a nefropatía clínica. En pacientes normotensos, la dosis será la máxima tolerada.
Desde el punto de vista terapéutico, la presencia de albuminuria representa un estadio evolutivo de la nefropatía diabética en la cual, el tratamiento es efectivo para prevenir la progresión de la enfermedad renal
Hipertensión arterial
Causas de hipertensión arterial en la adolescencia
La hipertensión primaria (HP), también conocida como hipertensión esencial, anteriormente considerada una enfermedad de la edad adulta, se ha vuelto cada vez más común en la población pediátrica en gran medida debido al incremento de las tasas de obesidad.
La hipertensión primaria, también conocida como hipertensión esencial, anteriormente considerada una enfermedad de la edad adulta, se ha vuelto cada vez más común en la población pediátrica en gran medida debido al incremento de las tasas de obesidad
Los antecedentes familiares de hipertensión de los padres están relacionados con un riesgo doblemente mayor de desarrollar hipertensión esencial en niños y adultos jóvenes(13). Esta asociación ha conducido a una extensa investigación para dilucidar la etiología genética subyacente de la HP. Los estudios familiares han demostrado que del 20 al 40% de los casos diagnosticados están determinados genéticamente.
Existe un creciente cuerpo de evidencia sobre la relación inversa entre el peso al nacer y la hipertensión en niños y adolescentes. Se ha observado una fuerte asociación entre pacientes con antecedentes de bajo peso al nacer y retraso del crecimiento intrauterino y se observa una relación más significativa cuando se realizan ajustes para el peso corporal actual(14). Las complicaciones a largo plazo de la hipertensión, como el accidente cerebrovascular, la insuficiencia cardíaca, el infarto de miocardio y la enfermedad renal son poco frecuentes en la población pediátrica.
La disfunción renal representa una forma de daño del órgano terminal relacionado con la hipertensión, que se manifiesta en forma de una reducción en la tasa de filtración glomerular y una excreción elevada de albúmina en la orina. La albuminuria se correlaciona bien con la progresión de la nefropatía. La hipertensión no controlada puede causar daño, también, a la vascularización retiniana.
En adolescentes se han descrito casos originados por causas de hipertensión arterial secundaria como síndrome de aorta media, arteritis de Takayasu, feocromocitoma, hipertensión de vena renal izquierda (Nutcraker) y se ha observado en pacientes operados de tetralogía de Fallot.
Hipertensión arterial y obesidad
La obesidad es una enfermedad crónica y compleja que suele iniciarse en la infancia y la adolescencia. En la actualidad es un importante y creciente problema de salud pública en dicho sector de la población. Su importancia radica, no sólo en su creciente prevalencia, sino también en las comorbilidades secundarias a ella. La obesidad está implicada en el desarrollo de diversas complicaciones de tipo metabólico y cardiovascular, típicas del adulto, pero que pueden estar presentes desde la adolescencia e incluso infancia. Entre ellas cabe destacar la resistencia a la insulina.
La resistencia a la insulina junto con la obesidad, constituyen la base fisiopatológica de la enfermedad cardiovascular. Ambas están relacionadas entre sí y con otros factores, tales como las alteraciones del metabolismo de los hidratos de carbono y de los lípidos y la hipertensión arterial, constituyendo lo que se conoce como Síndrome Metabólico.
En niños, existen escasos estudios al respecto pero los existentes, han demostrado que la presencia de obesidad y otros factores tales como hipertensión arterial, dislipemia o diabetes mellitus, se asocian a un engrosamiento de la pared arterial (medido como grosor de la intima-media de la carótida)(15). Estas lesiones son predictivas de riesgo cardiovascular en la edad adulta. No obstante, se objetiva su normalización si se actúa de forma temprana sobre ellas.
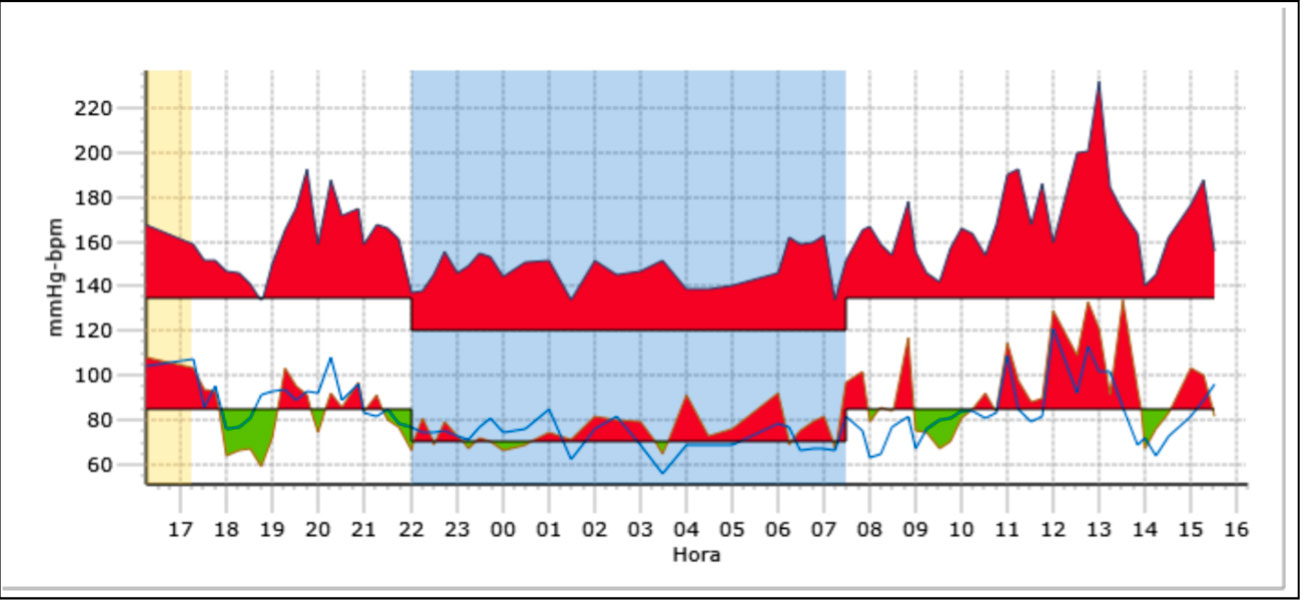
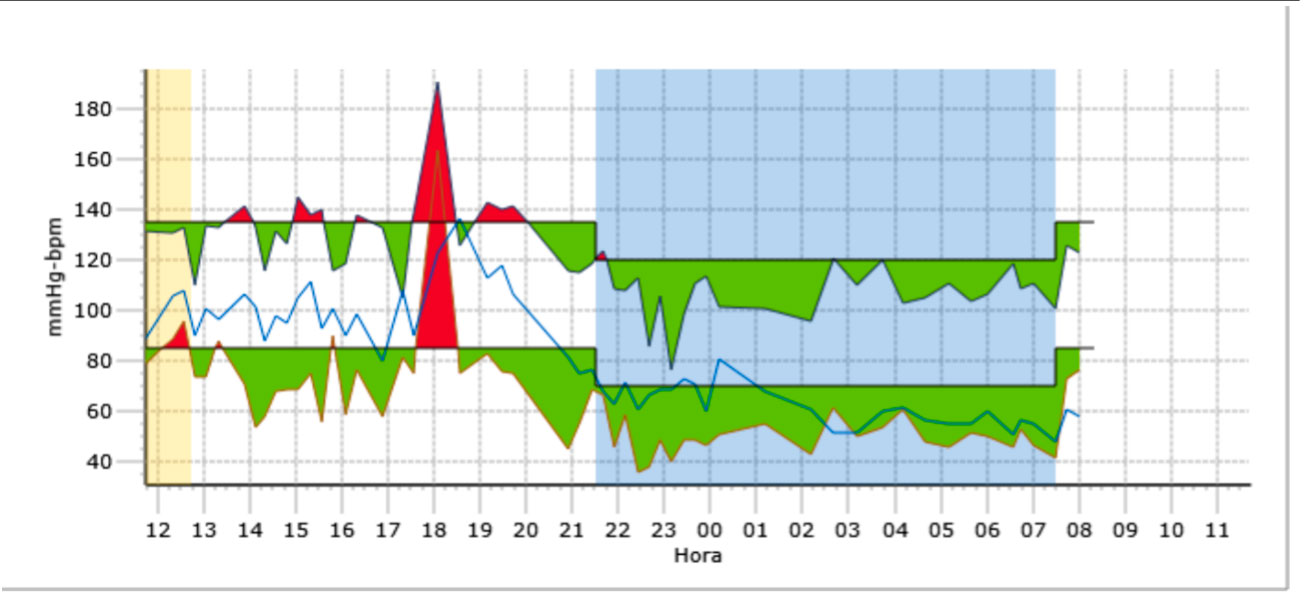
El uso de la monitorización ambulatoria de la presión arterial (MAPA) resulta muy útil. A lo largo de la última década, la MAPA ha emergido como una tecnología que soluciona varias de las limitaciones de las medidas casuales de la presión arterial en la consulta. Así, la valora múltiples veces durante un período de tiempo predefinido en el ambiente normal del paciente, tanto durante los períodos de vigilia como los de sueño, con lo cual se reduce la posibilidad de las elevaciones transitorias de PA producidas por el estrés. Esto permite evaluar no sólo las elevaciones casuales durante el día sino, también, las alteraciones en el patrón circadiano de la presión arterial a lo largo de las 24 horas.
En un estudio realizado por nuestro Grupo en una muestra constituida por 58 niños y 61 niñas obesos, observamos que la prevalencia global de hipertensión medida por MAPA era del 36%. El 14% eran hipertensos sistólicos diurnos, y el 33% hipertensos sistólicos nocturnos. De estos últimos, veinticinco de ellos (64%) sólo eran hipertensos sistólicos durante la noche, y el resto, también lo eran durante el día. Sólo cuatro pacientes eran hipertensos sistólicos diurnos de manera aislada. Ningún paciente presentó una hipertensión diastólica aislada diurna o nocturna. En el 47% del total de pacientes no se producía el descenso nocturno esperado de la PA sistólica. Esta pérdida del patrón circadiano de la PA dependía tanto del grado de obesidad como de la resistencia a la insulina (HOMA)(16).
Enfermedad de Gitelman
Los estudios de biología molecular han permitido distinguir claramente el síndrome de Bartter de una enfermedad con características similares, descrita en 1966 por Gitelman, Graham y Welt. Estos autores publicaron los datos clínicos de tres pacientes adultos, dos de ellos hermanos, afectos de hipopotasemia, hipomagnesemia y alcalosis metabólica. Durante muchos años, los pacientes con estas características fueron diagnosticados erróneamente de síndrome de Bartter. La presencia de hiperreninismo e hiperaldosteronismo, contribuyó a la confusión con el síndrome de Bartter clásico.
A finales de los años 80, la enfermedad de Gitelman o Hipomagnesemia-Hipopotasemia Familiar se identificó como una entidad distinta que se distinguía del síndrome de Bartter por la presencia de hipocalciuria, una capacidad de concentración renal prácticamente normal, una morfología glomerular renal normal en la biopsia renal y, a menudo, una sintomatología menos llamativa. El inicio de la clínica suele aparecer en la adolescencia, generalmente, con síntomas neuromusculares leves(17). El espectro de manifestaciones, no obstante, es amplio. Así, puede ser asintomática o expresarse con síntomas leves y, a veces, intermitentes (debilidad muscular, calambres, fatiga, poliuria, nicturia o dolor articular) o con síntomas más graves (tetania, convulsiones). Frecuentemente, ocurren parestesias, especialmente en la cara. La avidez por la sal es frecuente y los valores de presión arterial son más bajos que en la población general. El crecimiento no suele verse afectado, aunque se ha descrito fallo de medro y talla baja en una minoría de casos. La hipomagnesemia y la hipopotasemia prolongan la repolarización ventricular que predispone a que surjan arritmias graves. Por ello, los individuos con enfermedad de Gitelman deben evitar los deportes de competición dado que en pacientes con QT prolongado, la muerte súbita es precipitada por la actividad física. Algunos pacientes pueden tener únicamente síntomas en la edad adulta relacionados con la condrocalcinosis que causa hinchazón, calor local y sensibilidad incrementada en las articulaciones afectadas.
En pacientes con enfermedad de Gitelman la observación tanto, de que las anomalías electrolíticas se asemejaban a los efectos producidos por la administración crónica de tiazidas, como los resultados obtenidos en los estudios de aclaramientos, apuntaron a que el defecto tubular debía residir en el transporte distal de sodio y cloro sensible a tiazidas. En efecto, en 1996, se estableció que la enfermedad de Gitelman es producida por una reducción en el transporte de ClNa en el túbulo contorneado distal debido a la existencia de mutaciones en el gen SLC12A3 que codifica el cotransportador de ClNa sensible a tiazidas [renal thiazide-sensitive Na+-Cl− cotransporter NCC, thiazide sensitive contransporter, NCC], que se localiza en el lado luminal de las células del túbulo contorneado distal(18) (Figura 4).
En fisiología, Na+ y Cl– pasan desde la luz tubular al interior de la célula mediante el cotransportador de ClNa sensible a tiazidas, NCC. Mutaciones en el gen que codifica esa proteína causan la enfermedad de Gitelman. El Na+ sale de la célula mediante la Na+,K+,ATPasa. El Cl– sale de la célula mediante la acción del canal de cloro ClC-Kb. En la enfermedad de Gitelman, el descenso de actividad del cotransportador de ClNa NCC afecta el potencial de membrana necesario para la reabsorción apical de magnesio por parte del canal epitelial de magnesio TRPM6, que se localiza en la membrana apical del túbulo contorneado distal y en el borde en cepillo del duodeno. La hipomagnesemia de la enfermedad está causada, por tanto, por una pérdida urinaria e intestinal de magnesio. El mecanismo de la hipocalciuria observado en la enfermedad de Gitelman debe ser el mismo que ocurre con el uso de tiazidas. En ambos casos, el bajo contenido intracelular de sodio que se produce en las células del túbulo distal debido a la pérdida salina, favorece la salida de calcio de la célula y la entrada de sodio mediante la activación del intercambiador Na+/Ca++ basolateral, motivo por el que se reduce la calciuria. Al disminuir los niveles intracelulares de Ca++, difunde una mayor cantidad del ión hacia el interior de la célula por medio del canal catiónico selectivo de calcio apical TRPV5 (transient receptor potential cation channel subfamily V member 5; conocido anteriormente como epithelial Ca++ channel o ECaC1).
En 1996, se estableció que la enfermedad de Gitelman es producida por una reducción en el transporte de ClNa en el túbulo contorneado distal debido a la existencia de mutaciones en el gen SLC12A3 que codifica el cotransportador de ClNa sensible a tiazidas
Como en el síndrome de Bartter, el defecto de reabsorción de Na+ y Cl– en el túbulo contorneado distal determina una depleción de volumen moderada que estimula el sistema renina-angiotensina-aldosterona, favoreciendo de este modo la reabsorción de Na+ y la eliminación de K+ e H+ en los ductos colectores, dando lugar a la aparición de hipopotasemia y alcalosis metabólica. La contracción de volumen origina, además, un incremento en la reabsorción de bicarbonato en el túbulo proximal, lo que mantiene la alcalosis. La hipopotasemia se mantiene debido a la entrada de potasio en las células para equilibrar la salida de H+ de las mismas destinada, a su vez, a intentar equilibrar la alcalosis.
En todo caso, la pérdida salina es menor que el síndrome de Bartter, con lo que los niveles de renina y aldosterona no están tan elevados como en éste último (Figura 4).
Los pacientes muestran una respuesta natriurética anulada tras la administración de tiazidas. En cambio, la respuesta a la furosemida está conservada. El tratamiento consiste en la administración de sales de magnesio. La hipopotasemia se trata con el uso simultáneo de ClK y amiloride.
Malformaciones congénitas con pérdida de parénquima renal
Muchas malformaciones renales cursan con pérdida de parénquima, es decir, el número de nefronas es congénitamente reducido. Ejemplo de ellos son, por ejemplo, la agenesia renal unilateral, la displasia renal multiquistica, la hipoplasia e hipodisplasia renales, el reflujo vesicoureteral y la estenosis pieloureteral.
Para las formas más graves y más precoces de enfermedad renal, la mayor probabilidad de progresión a insuficiencia renal es en el primer año de vida, a los 5-6 años, o después del crecimiento acelerado en la adolescencia. Siendo estos dos tiempos de crecimiento global acelerado, esta observación ha llevado a algunos investigadores a proponer que el crecimiento somático en ausencia de crecimiento renal paralelo es la causa de dicha progresión a insuficiencia renal(19). Este hallazgo puede atribuirse a un cambio en la masa de nefronas disponible por unidad de peso corporal. Según el número de nefronas ausentes, la reducción del filtrado glomerular renal es más o menos marcado.
Para las formas más graves y más precoces de enfermedad renal, la mayor probabilidad de progresión a insuficiencia renal es en el primer año de vida, a los 5-6 años, o después del crecimiento acelerado en la adolescencia
La reducción de la masa renal bien es un factor de riesgo de desarrollo de enfermedad renal progresiva(20). Según la teoría de la hiperfiltración, la reducción de la masa nefrónica acarrea una sobrecarga funcional de las nefronas restantes que se verán sometidas a un estado permanente de hiperfiltración en un intento de mantener la función renal. Estos cambios inicialmente beneficiosos pueden condicionar a la larga un deterioro de la función renal. Los primeros datos sobre las lesiones escleróticas renales secundarias a la disminución de la masa renal fueron aportados por el estudio de modelos animales experimentales. Posteriormente, fueron descritas en humanos(21).
El bloqueo del SRA reduce la presión capilar glomerular e interfiere en los procesos inflamatorios y de fibrogénesis renales mediados por la angiotensina II (AII), obteniendo un efecto antiproteinúrico y renoprotector añadido a la acción antihipertensiva. Estudios experimentales recientes apuntan, además, a su acción reguladora de la función podocitaria(20). Los inhibidores de la enzima de conversión de la angiotensina (IECA) han demostrado influir favorablemente en la evolución del daño renal en animales de experimentación y humanos. Estos fármacos inhiben la ECA impidiendo la conversión de la angiotensina I a AII. Sin embargo, el bloqueo del SRA no es completo, ya que existen otras vías capaces de generar AII no dependientes de esta enzima. Los antagonistas de los receptores de la angiotensina AII (ARA) bloquean competitivamente el receptor AT-1 de la AII anulando completamente la acción mediada por este receptor. Además favorecen la unión de la AII circulante a los receptores AT-2 obteniendo un efecto vasodilatador y antiproliferativo.
Otras medidas destinadas a intentar reducir el ritmo de progresión del daño renal son el control de peso, de la hiperlipemia y de la presión arterial y una dieta reducida en proteínas.
Glomerulopatias
Las enfermedades glomerulares que con una mayor frecuencia precisan una biopsia renal en la adolescencia son las tres glomerulopatias que se mencionan a continuación.
Nefropatía IgA
Se trata de la glomerulonefritis primaria más prevalente en el mundo. Puede presentarse a cualquier edad, pero es más frecuente en la segunda y tercera década de la vida y en varones. No se conoce exactamente la patogénesis de la enfermedad pero se trata de una glomerulonefritis mediada por complejos inmunes o polímeros de IgA, los cuales se depositan a nivel del mesangio(22). Hay que tener en cuenta que un porcentaje sano de la población (3-16%) puede presentar depósitos mesangiales de IgA y, por tanto, los depósitos no inducen directamente el daño glomerular por lo que puede existir una cohorte grande de nefropatía IgA latente no diagnosticada en la población general. Además, se han documentado también depósitos de IgA en otras formas de glomerulonefritis (púrpura de Shönlein-Henoch, nefritis lúpica…). Existen casos familiares y esporádicos, teniendo estos últimos mejor pronóstico. No se ha logrado identificar el gen causal aunque sí algunos relacionados con una mayor susceptibilidad a padecer la enfermedad.
La nefropatía IgA es la forma más frecuente de glomerulonefritis. Cursa con episodios recurrentes de hematuria macroscópica, generalmente, 2-3 días después de padecer una infección respiratoria vírica o bacteriana (a diferencia de la glomerulonefritis aguda postinfecciosa que será habitualmente de 3 a 4 semanas después). Se puede acompañar de febrícula y/o dolor lumbar. Otra forma de presentación puede ser en forma de hematuria microscópica y proteinuria leve, detectándose en exámenes de orina rutinarios. Solo un porcentaje pequeño de pacientes puede debutar como un síndrome nefrótico o un síndrome nefrítico agudo con hematuria, hipertensión, edemas y daño renal agudo. Los factores clínicos de progresión de la enfermedad son el aumento de la creatinina, la hipertensión arterial y la proteinuria moderada persistente. Existen casos descritos de remisión espontánea, sobre todo en niños.
La nefropatía IgA es la forma más frecuente de glomerulonefritis. Cursa con episodios recurrentes de hematuria macroscópica, generalmente, 2-3 días después de padecer una infección respiratoria vírica o bacteriana (a diferencia de la glomerulonefritis aguda postin-fecciosa que será habitualmente de 3 a 4 semanas después)
El diagnóstico de esta enfermedad se sospechará por la clínica. A nivel analítico puede existir un aumento de los niveles séricos de IgA en un 8-15% de los niños y 30-50% de los adultos. Presentarán también un C3 y ASLO normal que se deberán solicitar para realizar el diagnóstico diferencial con otras entidades. La creatinina sérica se tiene que monitorizar, así como cuantificar la proteinuria, que es un indicador de progresión de la enfermedad. Finalmente, será la biopsia renal la que confirme el diagnóstico(23). Sin embargo, no se va a realizar de forma sistemática en todos los casos, se priorizará para aquellos con un síndrome nefrítico con proteinuria en rango nefrótico, una disminución progresiva de la función renal o proteinuria moderada persistente. Existen algunas enfermedades relacionadas con la nefropatía IgA como la cirrosis, la enfermedad celiaca y el VIH.
No existe un tratamiento curativo. En caso de proteinuria mantenida y creciente se utilizarán los IECAs- ARAII y, en el caso de progresión de la enfermedad, se podrán utilizar corticoides e inmunosupresores. En la insuficiencia renal terminal el tratamiento de elección será el trasplante renal.
La recurrencia postrasplante existe aunque la incidencia es baja (mayor si el trasplante es de donante vivo relacionado). Inicialmente se consideró una entidad benigna, pero el posterior seguimiento a largo plazo de los pacientes puso de manifiesto que entre un 20-50% de los adultos evolucionan a insuficiencia renal terminal.
Lupus eritematosos sistémico
El lupus eritematoso sistémico (LES) es una enfermedad autoinmune multisistémica compleja que cursa en brotes y que resulta de la interacción de factores ambientales, hormonales y genéticos. La presentación en la edad pediátrica tiene una incidencia del 15-20% del total de pacientes. La edad promedio de presentación es los 12 años; es más prevalente en mujeres y más grave que cuando se inicia en la edad adulta, con mayor afectación renal (60-80%), lo que determina el pronóstico.
La clínica es variable e impredecible. Se considera “la gran imitadora” dado que puede afectar a cualquier sistema (manifestaciones mucocutáneas, musculoesqueléticas, hematológicas, cardiovasculares, neuropsiquiátricas, oculares, etc). Es importante el conocimiento de esta enfermedad para poder sospecharla en una fase temprana.
Se considera “la gran imitadora” dado que puede afectar a cualquier sistema (manifestaciones mucocutáneas, musculoesqueléticas, hematológicas, cardiovasculares, neuropsiquiátricas, oculares, etc)
Ninguna prueba, por si misma, es diagnóstica de lupus. Individuos con autoanticuerpos positivos (ANA, anti-DNA) pueden estar sanos o bien presentar otras enfermedades diferentes al lupus. Existen unos criterios de clasificación según el Colegio Americano de Reumatología que constan de 11 apartados teniendo que poseer, al menos, cuatro criterios clínicos y/o de laboratorio para diagnosticar LES. Sin embargo, un paciente puede tardar años en cumplir dichos criterios. La afectación renal se debe estudiar en todos los pacientes con LES(24). Se deben evaluar proteinuria, sedimento urinario y función renal. En caso de afectación es preciso realizar una biopsia renal para clasificar la nefritis lúpica. Los datos clínicos y analíticos habituales no pueden predecir los hallazgos histológicos en un alto porcentaje de los casos. El diagnóstico anatomopatológico es clave para establecer el pronóstico y planificar el tratamiento. La OMS elaboró una clasificación en la que se describen seis formas anatomopatológicas de afectación renal.
El tratamiento será individualizado según las manifestaciones clínicas y la histología renal. El objetivo es preservar la función renal y prevenir la aparición de brotes. Los tratamientos incluyen corticoides, inmunosupresores, hidroxicloroquina (previene los brotes y aumenta la supervivencia a largo plazo), ARA II, AAS, calcio y vitamina D, estatinas, hipotensores y AINES, así como medidas preventivas como protección solar, pérdida de peso, ejercicio físico y evitar el tabaco y el estrés.
El pronóstico de los niños y adolescentes que reciben un tratamiento adecuado es generalmente bueno. El tratamiento temprano y agresivo en pacientes con síntomas severos se asocia a un curso favorable de la enfermedad. El factor pronóstico fundamental es la lesión histológica responsable. Las clases III y IV de nefropatía lúpica evolucionan a enfermedad renal crónica en un 25-40%.
Síndrome nefrótico
El síndrome nefrótico (SN) es la manifestación clínica de las alteraciones bioquímicas producidas por una lesión glomerular, concretamente del podocito (podocitopatía), que origina una alteración de la permeabilidad de la pared capilar glomerular y retención de sodio, dando origen a las características que lo definen: proteinuria mayor de 40 mg/m2/hora (50 mg/Kg/día), albuminemia inferior a 2.5 g/dl y edemas. Secundariamente, y por pérdida de otras proteínas además de la albúmina, asociará otras alteraciones como la dislipemia, la hipercoagulabilidad con tendencia a fenómenos tromboembólicos y la susceptibilidad a infecciones.
La etiología es desconocida. Se conoce que están involucrados mecanismos inmunológicos (alteraciones de los linfocitos T, B y factores de permeabilidad vascular) o genéticos (mutaciones en los genes de las proteínas podocitarias) de forma independiente o multifactorial. Es una entidad homogénea en su expresión clínica pero heterogénea en cuanto a su curso evolutivo, respuesta al tratamiento, pronóstico e histología renal. Este amplio espectro refleja los diferentes mecanismos moleculares implicados en la patogenia de la enfermedad(25).
El SN más frecuente es el denominado “idiopático” en el que la edad de aparición más habitual es entre los dos y los ocho años con una máxima incidencia entre los tres y los cinco años. Otros tipos de SN son el congénito (el producido en menores de un año de edad), SN genético aislado (habitualmente síndromes nefróticos corticorresistentes, con edad de presentación más tardía y con biopsias compatibles con glomeruloesclerosis segmentaria y focal) o sindrómico (asociado a síndromes) y SN secundarios a otras patologías específicas. Esta clasificación está sufriendo modificaciones debido a que se ha objetivado que existen mutaciones genéticas en la mayoría de los SN congénitos y familiares y en el 10-20 % de los SN resistentes esporádicos. En caso de un debut más tardío de la enfermedad, como es en la adolescencia, habrá que sospechar un SN de origen genético. Las mutaciones más frecuentes encontradas a esta edad se encuentran en los genes NPHS2 (autosómica recesiva y casos esporádicos), INF2 y TRPC6 (autosómica dominante)(26). Estos mismos genes, además de otros, están relacionados con la glomeruloesclerosis segmentaria y focal, que es muy probable que desarrollen estos pacientes durante su evolución.
Por otro lado, la clasificación clínica del SN se establece en función de la respuesta al tratamiento con corticoides: síndrome nefrótico corticosensible (resolución clínica y analítica), corticodependiente (más de dos recaídas al disminuir la dosis de corticoides o tras dos semanas después de la suspensión) y síndrome nefrótico corticorresistente (persiste el SN clínico y/o bioquímico a pesar de tratamiento durante 4-6 semanas). Estas clasificaciones tienen un gran valor pronóstico y condicionarán la indicación de un posible tratamiento inmunosupresor.
La histología más frecuente en las biopsias son lesiones mínimas o riñón ópticamente normal, glomeruloesclerosis segmentaria focal (GESF) y glomerulonefritis proliferativa mesangial. Las dos últimas presentan peor respuesta y pronóstico a largo plazo. La indicación para la realización de una biopsia renal en esta entidad es: edad de debut por debajo de los 12 meses de edad, SN con inicio de enfermedad en la adolescencia, SN de carácter familiar, resistencia a corticoides, deterioro del filtrado glomerular o signos de SN secundario a una enfermedad sistémica o infecciosa y cambio desfavorable de la respuesta al tratamiento con corticoides como la evolución de corticosensibilidad/dependencia a corticorresistencia.
El tratamiento inicial son los corticoides y la respuesta a los mismos y la evolución determinarán la necesidad de una terapia alternativa con inmunosupresores como ciclofosfamida, ciclosporina, micofenolato, tacrolimus y, en los últimos años, también rituximab. La mayoría de los niños con SN idiopático responde al tratamiento pero alrededor del 20% son corticorresistentes. Más de dos tercios presenta recaídas de la enfermedad en los primeros dos meses y hasta el 60% tienen dependencia de los corticoides. Se considera enfermedad en remisión completa tras 7-10 años sin recaídas. La mayoría de los niños con SN corticonsensible e histología de cambios mínimos alcanzan la edad adulta en remisión completa. Sin embargo, hasta en una cuarta parte puede persistir la actividad después de los 18 años y hasta un 40% puede presentar alguna recaída en la edad adulta. La mayoría de los niños que recaen continúan siendo corticosensibles y mantienen una función renal normal. Por contra, el síndrome nefrótico corticorresistente está asociado en un 30-50 % a una evolución a enfermedad renal terminal en cinco años, si no se logra controlar.
La microscopía óptica no mostró alteraciones y en la inmunofluorescencia directa se observaron muy discretos depósitos granulares de IgM sin depósitos de IgG, IgA, C3, C1q o fibrinógeno. En la microscopía electrónica se confirmó una fusión de pedicelos (podocitopatía). En este paciente, aunque inicialmente la biopsia se trate de lesiones mínimas, lo más probable es que durante su evolución desarrolle una glomeruloesclerosis segmentaria y focal.
Tablas y figuras
Tabla I. Infecciones del tracto urinario
|
Microorganismos
más frecuentes
|
Menos frecuentes
|
Raros
|
|
Cistitis aguda
|
E. coli
|
Proteus
Klebsiella
S. saprophyticus
|
Enterococo, Pseudomona,
Corynebacterium urealyticum,
Morganella, Citrobacter,
Enterobacter, Serratia,
Salmonella, Shigella
|
|
Pielonefritis aguda
|
E. coli
Proteus
|
Klebsiella, Morganella,
Citrobacter, Enterobacter,
Serratia, Pseudomona,
Enterococo
|
S. aureus,
S. saprophyticus,
Salmonella
|
Modificada de González Monte E. Infecciones del tracto urinario. Lorenzo V, López Gómez JM, eds. Nefrología al día. Actualizado en junio de 2018.
Tabla II. Cistitis aguda en niños mayores y adolescentes
|
Hallazgo patológico
|
S (%)
|
E (%)
|
VPP/VPN (%)
|
|
Tira reactiva
|
Esterasa leucocitaria +
Nitritos +
Esterasa leucocitaria y nitritos +
|
85
50
72
|
78
98
96
|
45/89
97/90
53/93
|
|
Sedimento urinario
|
>10 leucocitos/ campo en orina no centrifugada ó
> 5 leucocitos/campo en orina centrifugada
|
73
|
81
|
|
Modificada de Caballero García M, López Lledó S. Cistitis aguda en niños mayores y adolescentes Guía-ABE. Infecciones en Pediatría. Guía rápida para la selección del tratamiento antimicrobiano empírico (v.1/2011).
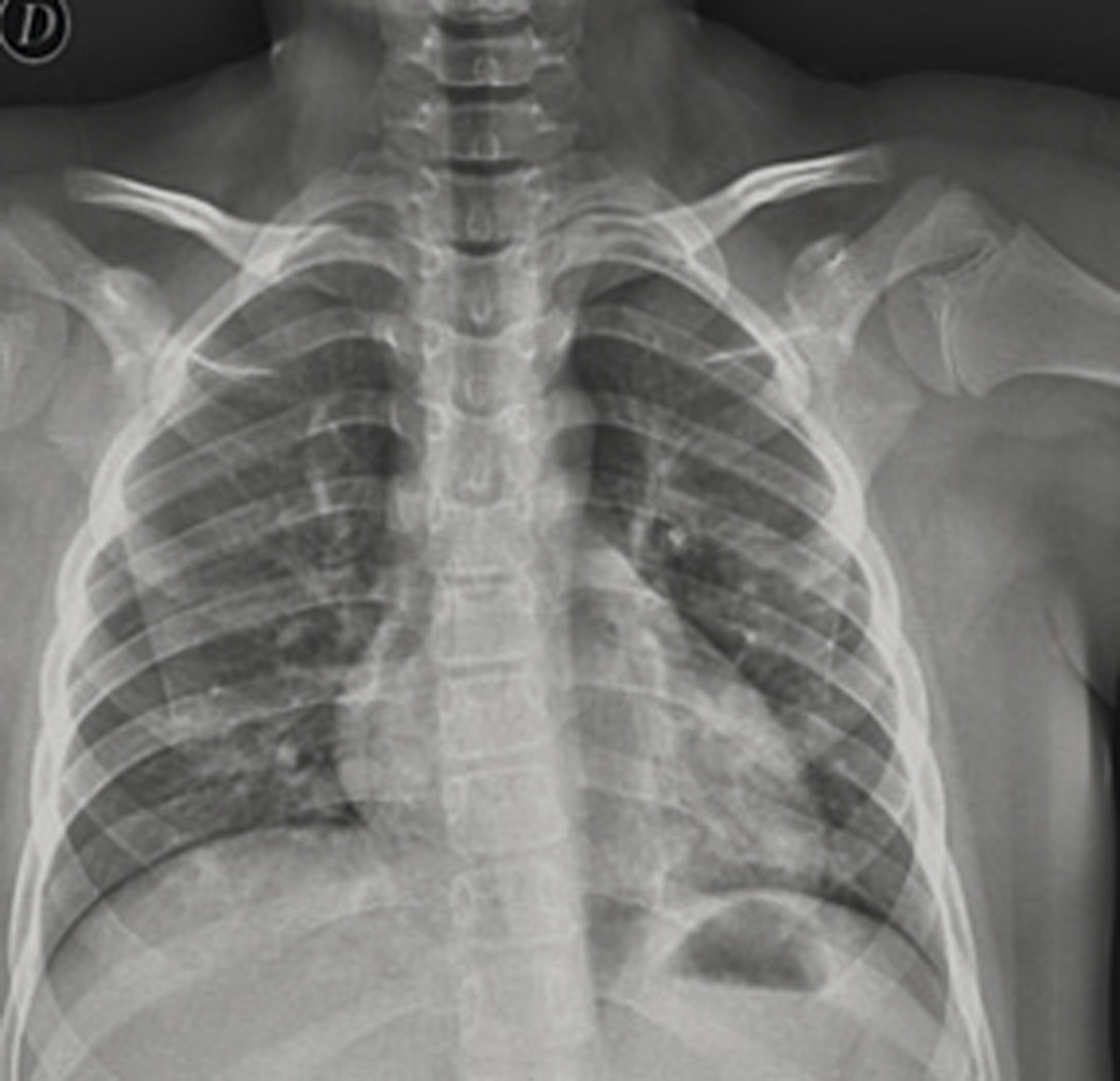
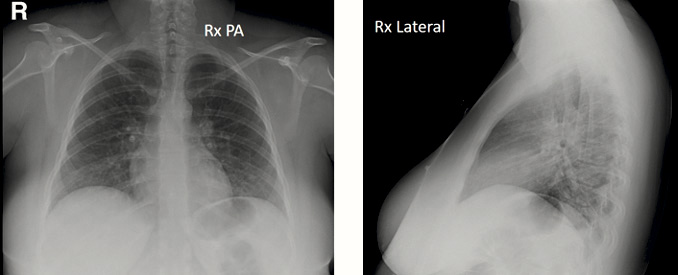
Figura 1. Imagen ecográfica de un cálculo ubicado en la unión vesicoureteral
Figura 2. Litiasis fosfática infecciosa
(Fosfato amónico magnésico o estruvita)

La infección bacteriana prolongada del tracto urinario suele ser la causa más común de este tipo de litiasis. Los gérmenes ureolíticos (Proteus, Klebsiella, Pseudomona, Ureaplasma…) suelen provocar una notable elevación del pH urinario (pH> 7) y de la concentración urinaria de amonio, que conjuntamente favorecen la precipitación de fosfato amónico magnésico y de hidroxiapatita (fosfato de calcio). Detalle de un cristal de estruvita en cuyas caras se observan las marcas en “Y” que permiten su rápida identificación junto pequeñas zonas de esferulitos de hidroxiapatita (cortesía del Dr. Félix Grases. Laboratorio de Investigación en Litiasis Renal, Instituto Universitario de Investigación en Ciencias de la Salud, Universidad de las Islas Baleares, Palma de Mallorca, España).
Figura 3. Vista general y detalle al microscopio electrónico de barrido del interior del “cálculo” de carbonato cálcico

Paciente que a los 14 años expulsó tres cálculos. En el estudio metabólico se observó hipocitraturia. Vista general y detalle al microscopio electrónico de barrido del interior del “cálculo” de carbonato cálcico. La estructura no se corresponde con la típica de un cálculo renal con una morfología correspondiente a una estructura de origen vegetal, por lo que podría tratarse de una semilla (Dr. Grases).
Figura 4. Mecanismos de transporte en el túbulo contorneado distal
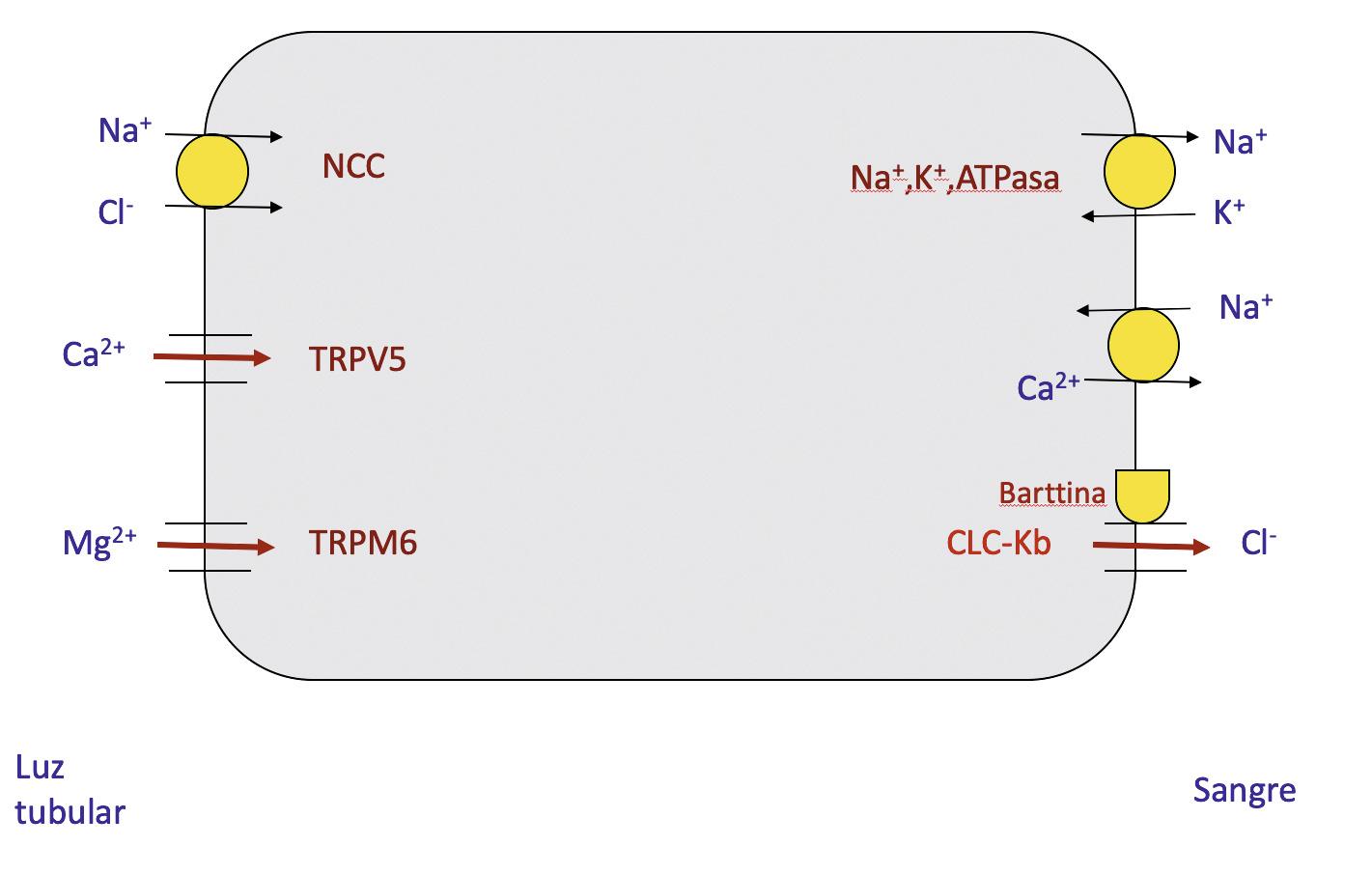
En fisiología, Na+ y Cl– pasan desde la luz tubular al interior de la célula mediante el cotransportador de ClNa sensible a tiazidas, NCC. Mutaciones en el gen que codifica esa proteína causan la enfermedad de Gitelman. El Na+ sale de la célula mediante la Na+,K+,ATPasa. El Cl– sale de la célula mediante la acción del canal de cloro ClC-Kb. En la enfermedad de Gitelman, el descenso de actividad del cotransportador de ClNa NCC afecta el potencial de membrana necesario para la reabsorción apical de magnesio por parte del canal epitelial de magnesio TRPM6, que se localiza en la membrana apical del túbulo contorneado distal y en el borde en cepillo del duodeno. La hipomagnesemia de la enfermedad está causada, por tanto, por una pérdida urinaria e intestinal de magnesio. El mecanismo de la hipocalciuria observado en la enfermedad de Gitelman debe ser el mismo que ocurre con el uso de tiazidas. En ambos casos, el bajo contenido intracelular de sodio que se produce en las células del túbulo distal debido a la pérdida salina, favorece la salida de calcio de la célula y la entrada de sodio mediante la activación del intercambiador Na+/Ca++ basolateral, motivo por el que se reduce la calciuria. Al disminuir los niveles intracelulares de Ca++, difunde una mayor cantidad del ión hacia el interior de la célula por medio del canal catiónico selectivo de calcio apical TRPV5 (transient receptor potential cation channel subfamily V member 5; conocido anteriormente como epithelial Ca++ channel o ECaC1).
Figura 5. Biopsia renal de un paciente adolescente con un síndrome nefrótico corticorresistente
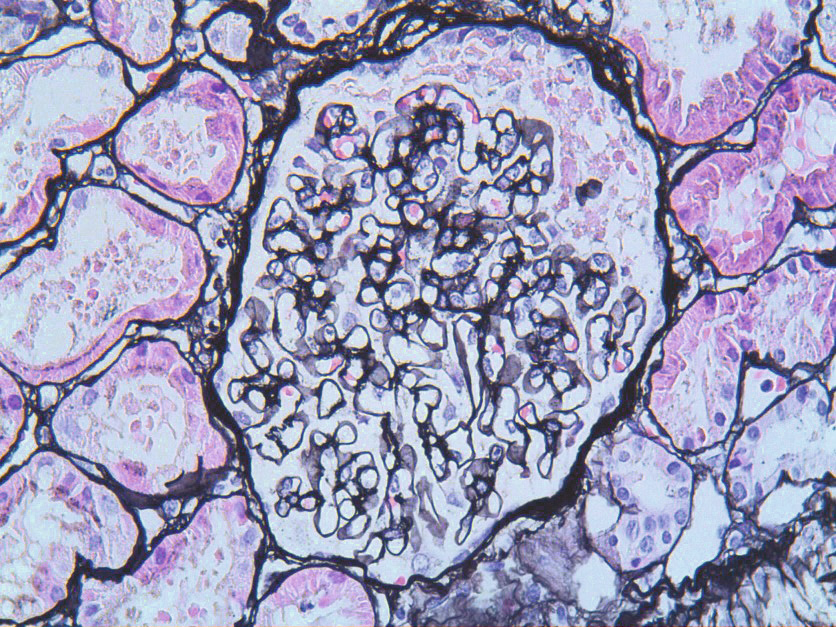
La microscopía óptica no mostró alteraciones y en la inmunofluorescencia directa se observaron muy discretos depósitos granulares de IgM sin depósitos de IgG, IgA, C3, C1q o fibrinógeno. En la microscopía electrónica se confirmó una fusión de pedicelos (podocitopatía). En este paciente, aunque inicialmente la biopsia se trate de lesiones mínimas, lo más probable es que durante su evolución desarrolle una glomeruloesclerosis segmentaria y focal.
Bibliografía
1. Shaikh N, Craig J, Rovers M, Da Dalt L, Gardikis S, Hoberman A et al. Identification of children and adolescents at risk for renal scarring after a first urinary tract infection. JAMA Pediatrics 2014; 168:893-900.
2. Pailhoriès H, Cassisa V, Chenouard R, Kempf M, Eveillard M, Lemarié C. Staphylococcus saprophyticus: Which beta-lactam? Int J Infect Dis 2017; 65:63-66.
3. Keating GM. Fosfomycin trometamol: a review of its use as a single-dose oral treatment for patients with acute lower urinary tract infections and pregnant women with asymptomatic bacteriuria. Drugs 2013; 73:1951-1966.
4. Molina Gil-Bermejo J, Cabello V, Campoy Martínez P, Barrera Chacón J. Cistitis y pielonefritis aguda. Guías para el diagnóstico y tratamiento de las enfermedades infecciosas. Hospital Universitario Virgen del Rocío, 2018.
5. Arias Vega R, Pérula de Torres LA, Jiménez García C, Carrasco Valiente J, Requena Tapia MJ, Cano Castiñeira R et al. Comorbidity and socio-demographic factors associated with renal lithiasis in persons aged 40 to 65: A cross-sectional study. Med Clin (Barc) 2017; 149:383-390.
6. Grases Freixedas F, Costa-Bauzá A. Mecanismos de la formación de los cálculos renales. En: Nefrología Pediátrica (2ª ed.). García Nieto V, Santos Rodríguez F, Rodríguez-Iturbe B, eds. Madrid: Aula Médica 2006, pp. 917-92.
7. García Nieto VM, Pérez Bastida XI, Salvador Cañibano M, García Rodríguez VE, Monge Zamorano M, Luis Yanes MI. Quantification of the risk of urinary calcium stone formation in the urine collected at 2 times of the day in a group of children studied to rule out prelithiasis. Nefrología 2018; 38:267-272.
8. Moore ES. Hypercalciuria in children. Contr Nephrol (Vol. 27). Basel:Karger 1981; 20-32.
9. Diabetes Control and Complications Trial/epidemiology of Diabetes Interventions and Complications (DCCT/EDIC) Research Group, Nathan DM, Zinman B, Cleary PA, Backlund JY, Genuth S, Miller R et al.. Modern-day clinical course of type 1 diabetes mellitus after 30 years’ duration: the diabetes control and complications trial/epidemiology of diabetes interventions and complications and Pitssburgh epidemiology of diabetes complications experience (1983-2005). Arch Intern Med 2009; 169:1307-1316.
10. Lurbe E, Redon J, Kesani A, Pascual JM, Tacons J, Alvarez V et al. Increase in nocturnal blood pressure and progression to microalbuminuria in type 1 diabetes. N Engl J Med 2002; 347:797-805.
11. Commissariat PV, Volkening LK, Guo Z, ElBach JL, Butler DA, Laffel LM. Associations between major life events and adherence, glycemic control, and psychosocial characteristics in teens with type 1 diabetes. Pediatr Diabetes 2018; 19:85-91.
12. Perkins BA, Ficociello LH, Kristen HS, Finkelstein DM, Warram JH, Krowlewski AS. Regression of microalbuminuria in type 1 diabetes. N Engl J Med 2003; 348:2285-2293.
13. Goldstein IB, Shapiro D, Weiss RE. How family history and risk factors for hypertension relate to ambulatory blood pressure in healthy adults. J Hypertens 2008; 26:276-283.
14. Primatesta P, Falaschetti E, Poulter NR. Birth weight and blood pressure in childhood: results from the Health Survey for England. Hypertension 2005; 45:75-79.
15. Woo KS, Chook P, Yu CW, Sung RY, Qiao M, Leung SS et al. Overweight in children is associated with arterial endothelial dysfunction and intima-media thickening. Int J Obesity 2004; 28:852-857.
16. Ruiz Pons M, García Nieto V, González García M, García Mérida M, Valenzuela Hernández C, Aguirre-Jaime A. Reduced nocturnal systolic blood pressure dip in obese children. Nefrología 2008; 28:517-524.
17. Naesens M, Steels P, Verberckmoes R, Vanrenterghem Y, Kuypers D. Bartter’s and Gitelman’s syndromes: from gene to clinic. Nephron Physiol 2004; 96:65-78.
18. Simon DB, Nelson-Williams C, Bia MJ, Ellison D, Karet FE, Molina AM et al. Gitelman’s variant of Bartter’s syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nat Genet 1996; 12:24-30.
19. Gonzalez Celedón C, Bitsori M, Tullus K. Progression of chronic renal failure in children with dysplastic kidneys. Pediatr Nephrol 2007; 7:1014-1020.
20. Brenner BM, Mackenzie HS. Nephron mass as a risk factor for progression of renal disease. Kidney Int 1997; S63:S124-S127.
21. Reiser J, Gersdorff G, Simons M, Schwarz K, Faul C, Giardino L, Heider T, Loos M, Mundel P. Novel concepts in understanding and management of glomerular proteinuria. Nephrol Dial Transplant 2002; 17:951-955.
22. Pillai U, Balabhadraputani K, Bhat Z. Immunoglobulin A nephropathy: A review of current literature on emerging pathophysiology. Am J Med Sci 2014; 347:249-253.
23. Coppo R. Clinical and histological risk factors for progression of IgA nephropathy: an update in children, young and adult patients. J Nephrol 2017; 30:339-346.
24. Ruiz Irastorza G, Espinosa G, Frutos MA, Jiménez Alonso J, Praga M, Pallarés L et al. Diagnóstico y tratamiento de la nefritis lúpica. Documento de consenso del Grupo de Enfermedades Autoinmunes Sistémicas (GEAS) de la Sociedad Española de Medicina Interna (SEMI) y de la Sociedad Española de Nefrología (SEN). Nefrología 2012; 32:1-35.
25. Wang CS, Greenbaum LA. Nephrotic syndrome. Pediatr Clin North Am 2019; 66:73-85.
26. Santin S, Bullich G, Tazón-Vega B, García-Maset R, Giménez I, Silva I. Clinical utility of genetic testing in children and adults with steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol 2011; 6:1139-1148.