Controversias en el tratamiento de la talla baja idiopática (TBI)
Controversias en el tratamiento de la talla baja idiopática (TBI)
J. Pozo Román.
Médico Adjunto del Servicio de Endocrinología Pediátrica del Hospital Infantil Universitario Niño Jesús de Madrid y Profesor Asociado de Pediatría de la Universidad Autónoma de Madrid.
Adolescere 2019; VII (2): 22-29
Resumen
|
Pocos temas en Endocrinología pediátrica han causado más discusión y diferentes opiniones que la talla baja idiopática (TBI), especialmente en lo referente a su definición, metodología diagnóstica y actitud terapéutica. A lo largo del artículo, se analiza: el concepto de TBI, controvertido, artificial y heterogéneo, fruto de nuestra incapacidad para llegar a un diagnóstico etiopatogénico; su diagnóstico, fruto de la exclusión de causas patológicas de talla baja; la dificultad en establecer los límites del esfuerzo diagnóstico; y por último, la controvertida actitud de tratar o no tratar, fundamentalmente mediante el empleo off-label de la hormona de crecimiento. Palabras clave: Talla baja; Talla baja idiopática; Hormona de crecimiento; Inhibidores de la aromatasa |
Abstract
|
There are few issues in pediatric endocrinology that have lead to more discussion and different opinions than idiopathic short stature (ISS), especially regarding its definition, diagnostic methodology and therapeutic attitude. This article analyzes: the concept of ISS, controversial, artificial and heterogeneous, the result of our inability to reach an etiopathogenic diagnosis; its diagnosis, a result of excluding pathological causes of short stature; the difficulty in establishing the limits of diagnostic effort; and finally, the controversial attitude of treating (or not), mainly with off-label growth hormone. Short stature; Idiopathic short stature; Growth hormone; Aromatase inhibitors. |
Introducción
La utilización terapéutica de la hormona de crecimiento (hGH) se inicio en 1958 y hasta 1985 estuvo limitada a pacientes gravemente deficientes de GH; ya que, la hGH es una hormona específica de especie y se obtenía por extracción a partir de hipófisis de cadáveres (pit-hGH), lo que reducía dramáticamente su disponibilidad(1). En 1985, su utilización se detuvo abruptamente, cuando se puso de manifiesto la asociación de determinados lotes de pit-hGH con el desarrollo de la enfermedad de Creutzfeldt-Jakob. Prácticamente, de forma simultánea, se comercializó la hGH biosintética (rhGH), obtenida por bioingeniería genética, que no solo eliminaba el riesgo de transmisión de enfermedades por virus lentos, sino que estaba disponible en cualquier cantidad, solo limitada por su costo, lo que terminó expandiendo su uso a otras indicaciones y haciendo desaparecer del mercado la pit-hGH.
La gran expansión terapéutica de la rhGH a partir de 1985 se produjo no sólo como terapia sustitutiva en pacientes deficitarios, incluyendo desde las formas más graves a las menos graves (formas no clásicas: deficiencias parciales de GH, disfunción neurosecretora de GH-DNSGH-, etc.), sino también como terapia farmacológica, no sustitutiva, en prácticamente cualquier forma de talla baja. Este proceso expansivo tenía su base en una serie de premisas no bien demostradas y que hoy en día siguen siendo cuestionadas por la escasez de evidencia científica sólida que las sostenga, como son que:
- La talla baja supone un hándicap en la vida de un sujeto que requiere un adecuado tratamiento;
- La rhGH es bien tolerada, incluso a dosis altas/suprafisiológicas en niños sin deficiencia de GH; y
- El aumento de talla inducido por la rhGH mejora la calidad de vida.
Durante los años posteriores a 1985, en función de los resultados obtenidos en los diferentes ensayos clínicos y de la eficacia y seguridad del tratamiento con rhGH, se fueron aprobando y consolidando nuevas indicaciones para el empleo de la rhGH en los diferentes países, incluyendo en el caso de los EE.UU., pero no en el de Europa, la talla baja idiopática (TBI). La aprobación de estas indicaciones en formas de talla baja no dependientes de rhGH (síndrome de Turner, insuficiencia renal, pequeño para edad gestacional, haploinsuficiencia del gen SHOX, síndrome de Prader-Willi), validaba el hecho de que si la rhGH incrementaba la talla en niños sin deficiencia de GH, la etiología de la talla baja no era relevante a la hora de tratar con rhGH.
Concepto y epidemiología
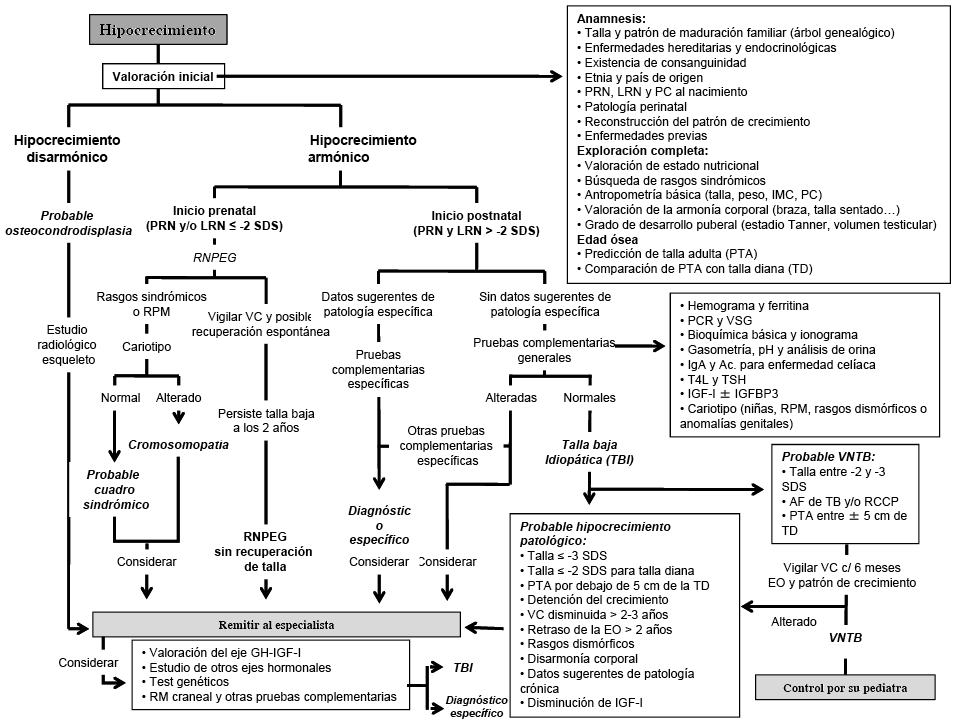
El concepto de “talla baja idiopática” (TBI), tal como lo entendemos hoy día y como ha sido asumido por la comunidad internacional, fue propuesto inicialmente por Allen en 1996(2). Su definición actual es el resultado del consenso entre expertos de las sociedades Americana (Lawson Wilkins Pediatric Endocrine Society) y Europea de Endocrinología Pediátrica (European Society for Paediatric Endocrinology), así como de la Growth Hormone Research Society, reunidos en Santa Mónica (California) en octubre de 2007(3-5). En esta reunión, se definió la TBI, como: una condición en la que la talla de un individuo se encuentra más de 2 SDS (percentil 2,3) por debajo de la media para su edad, sexo y grupo de población, sin evidencia de anomalías sistémicas, endocrinas, nutricionales o cromosómicas. Esta definición, según establecía el propio consenso, incluiría a los niños con variantes normales de talla baja [VNTB: talla baja familiar (TBF), retraso constitucional del crecimiento y de la pubertad (RCCP) y mezcla de ambos cuadros clínicos] y excluiría, específicamente, niños con: peso y/o talla baja para su edad gestacional, fenotipo dismórfico o disarmónico (displasias óseas, síndromes), trastorno psiquiátrico o emocional grave u otras causas claramente identificables de talla baja (enfermedad celíaca, enfermedad inflamatoria intestinal, malnutrición, deficiencia o insensibilidad a la GH, hipotiroidismo, síndrome de Cushing, etc.). Las VNTB serían hipocrecimientos armónicos de inicio postnatal que resultarían de la variabilidad normal tanto de la talla como del ritmo madurativo que existe en la especie humana.
La talla baja idiopática se define cuando la talla de un individuo se encuentra más de 2 SDS (percentil 2,3) por debajo de la media para su edad, sexo y grupo de población, sin evidencia de anomalías sistémicas, endocrinas, nutricionales o cromosómicas
Los avances fisiopatológicos más relevantes de los últimos años, en lo referente al crecimiento normal y patológico, han venido de la mano de la genética(6). Las variaciones normales en la talla son debidas en gran medida (80%) a factores genéticos, hereditarios; mientras que, el 20% restante se deberían a factores ambientales que contribuyen a la diferencia de talla entre las poblaciones y que serían responsables de la evolución secular de la talla a través de generaciones. La talla es un rasgo poligénico y los estudios amplios del genoma (GWAS) llevados a cabo en los últimos años han permitido identificar más de 600 variantes génicas comunes (presentes en ≥ 5%) en la población general, cada una de las cuales tendría una pequeña repercusión sobre la talla. Se calcula que los efectos aditivos de estas variantes comunes, la mayoría de ellas todavía por identificar, supondrían, al menos, la mitad de la variabilidad en la talla adulta de una población (Daubert, 2014). La causa del resto de la variabilidad de base genética no ha sido identificada, pero podría tratarse de variantes genéticas menos frecuentes, variante raras o interacciones entre variantes genéticas. Los efectos combinados de cientos de genes explicarían mucha de la variación dentro del rango normal de talla, pero la presencia de variantes más raras jugaría un papel prominente a medida que la talla baja se hace más extrema (formas de talla baja monogénica).
La TBI se subclasificaría según criterios auxológicos en:
- TBI familiar (FSS: familial short stature), cuando la talla es inferior a -2 SDS, pero adecuada a su contexto familiar.
- TBI no familiar (NFSS: non-familial short stature), cuando la talla es inferior a -2 SDS y se encuentra por debajo del rango normal para su talla familiar.
En ambos casos, el consenso contempla considerar también si la edad ósea (EO) se encuentra o no retrasada, como potencial indicador de la presencia/ausencia de retraso puberal y de la expectativa de una mejor talla adulta en los paciente con retraso de la EO(5).
Cuando en EE.UU. se aprobó, en junio de 2003, la utilización de la rhGH en el tratamiento de la TBI, la FDA (Food and Drug Administration) incluyó unos criterios más restrictivos para la indicación del tratamiento: una talla inferior a -2,25 SDS (percentil 1,2) y un ritmo de crecimiento que hiciera improbable alcanzar una talla adulta dentro del rango normal, entendiendo por tal: una talla inferior a 63 pulgadas (160 cm) en varones, y a 59 pulgadas (150 cm) en mujeres(5).
En Europa, la indicación de tratamiento no ha sido aprobada por la EMA (European Medicines Agency), debido a diferencias de opinión en relación con la eficacia, beneficio clínico y coste-efectividad; no obstante, en España, puede administrarse como un medicamento “fuera de indicación” (medicamento off-label)(7). De hecho, se calcula que la utilización off-label de la rHGH en España puede llegar a ser del 35% en algunas CC.AA. El Real Decreto 1015/2009, por el que se regula la utilización de medicamentos en situaciones especiales, define los medicamentos off-label o su “uso fuera de indicación”, como: “aquellos medicamentos utilizados en condiciones distintas de la incluidas en la ficha técnica autorizada”. La utilización de un medicamento off-label se encuentra en un terreno de grises, ya que no está contraindicado, pero su indicación no se encuentra recogida como habitual en la ficha técnica, bien por falta de estudios que lo avalen, o bien porque la legislación nacional de ese medicamento no la contempla, aunque pueda tener su aprobación en otros ámbitos internacionales. La legislación contempla en el uso off label de un medicamento una serie de condiciones:
- Carácter excepcional con falta de alternativas terapéuticas autorizadas;
- Necesidad de justificar en la historia clínica la necesidad de su empleo (cierta evidencia científica que avale su uso);
- Obligación de informar de los posibles beneficios y potenciales riesgos; y
- Necesidad de obtener un consentimiento del paciente o sus representantes legales.
El concepto de TBI es controvertido, artificial y heterogéneo; ya que, aunque se trataría teóricamente de niños con talla baja sin patología, lo cierto es que incluye situaciones normales y patológicas, cuyo único denominador común es nuestra incapacidad para alcanzar un diagnóstico etiopatogénico.
Se estima que, aproximadamente, el 80% de los niños que consultan por talla baja podrían ser diagnosticados de TBI. La inmensa mayoría de ellos (80-85%) corresponderían a VNTB y un pequeño porcentaje (15-20%) a patologías en las que, por desconocimiento o dificultad diagnóstica, no se llega a alcanzar un diagnóstico, como sería el caso de: hipocrecimientos nutricionales, hipocrecimientos psicosociales, alteraciones infrecuentes o menores en el eje GH-IGFs (deficiencia parcial idiopática de GH, mutaciones en el receptor de GH, mutaciones en ALS…), así como osteocondrodisplasias, cuadros sindrómicos o alteraciones genéticas de escasa expresividad clínica, entre otras posibles causas.
Se estima que, aproximadamente, el 80% de los niños que consultan por talla baja podrían ser diagnosticados de TBI. La inmensa mayoría de ellos (80-85%) corresponderían a VNTB y un pequeño porcentaje (15-20%) a patologías en las que no se llega a alcanzar un diagnóstico
Para algunos autores, el término “idiopático” implica que la etiología de la talla baja es desconocida y debería ser establecida descartando previamente las VNTB (TBF y RCCP) y especialmente el RCCP, ya que muchos de estos pacientes, pero no todos, alcanzan una talla adulta dentro del rango de la normalidad. Desgraciadamente, la EO no permite establecer de manera fiable, el momento de inicio puberal y el diagnóstico de RCCP debe retrasarse hasta que la pubertad se inicia, lo que dificulta el diagnóstico del RCCP.
Diagnóstico
El concepto de TBI como una talla inferior a -2 SDS para la edad, sexo y población, en la que se ha descartado patología (diagnóstico de exclusión), conlleva que se trataría de sujetos sanos con talla baja, pero no establece hasta dónde debe llegar el esfuerzo diagnóstico para excluir causas patológicas de talla baja. La anamnesis, una cuidadosa exploración general, auxológica y dismorfológica, así como unas pruebas complementarias básicas (hemograma, VSG, bioquímica básica, gasometría, metabolismo P-Ca, función tiroidea, IGF-I, IGFBP-3, marcadores de celiaquía y una edad ósea) son la base del estudio inicial. Mayores estudios en el eje de la GH pueden ser necesarios dependiendo de los niveles séricos de IGF-I e IGFBP-3 y de las características del patrón de crecimiento.
El eje GH-IGF1 es el eje hormonal más importante en la regulación del crecimiento lineal y alrededor del 25% de los pacientes con TBI muestran niveles disminuidos o en el rango bajo de la normalidad de IGF-I y, en menor medida de IGFBP-3, lo que sugiere un cierto grado de deficiencia o resistencia a la GH. Las dificultades en el diagnóstico de la deficiencia idiopática de GH (DIGH) y el hecho de que un alto porcentaje de estas deficiencias se corrijan espontáneamente tras la pubertad sugiere que, al menos, algunos de los pacientes diagnosticados de DIGH no serían verdaderas deficiencias sino formas de TBI, probablemente VNTB o hipocrecimientos nutricionales, psicosociales o sindrómicos con escasa expresividad clínica (síndrome de Noonan u otras rasopatías…). Por el contrario, algunas mutaciones en genes relacionados con el eje GH-IGF podrían condicionar sutiles cambios en la secreción de GH o en alguno de los componentes del eje distal de la GH, que podrían confundirse con VNTB(6,8,9). Así, por ejemplo, mutaciones en el gen de la GH (GH biológicamente inactiva), mutaciones en heterocigosis en la porción extracelular del receptor de GH, de herencia AD, anomalías en la transmisión de la señal de GH al núcleo (mutaciones en STAT5B…) o anomalías en biodisponibilidad del IGF-I como resultado de mutaciones en homocigosis o heterocigosis (portadores) de la subunidad ácido lábil (ALS) del complejo trimolecular que forman IGF1, IGFBP3 y ALS, pueden dar lugar a cuadros clínicos de talla baja más o menos grave con niveles más o menos disminuidos de IGF1 e IGFBP3. Más recientemente, se han descrito mutaciones en homocigosis en la papalisina 2 (PAPP-A2), la enzima proteolítica que rompe el complejo trimolecular IGF-IGFBP-3-ALS, lo que determinaría una falta de liberación de IGFs a los tejidos y que cursa con niveles séricos elevados de los tres componente del complejo y una afectación leve-moderada de la talla. Nuevos componentes del eje distal de la GH, como la staniocalcinas, que regulan la actividad de la PAPPA-2, podrían ser otros posibles candidatos a alteraciones del eje GH-IGF1, susceptibles de ser confundidas con formas de TBI.
Dentro del esfuerzo diagnóstico habitual en niños con TBI, un cariotipo debe realizarse siempre en niñas y, probablemente, también en varones (mosaicismos 45X/46,XY con talla baja aislada), así como un estudio radiográfico del esqueleto ante la mínima sospecha de displasia ósea.
Se han descrito más de 400 formas de displasia ósea, cuya manifestación principal, además de la talla baja, es la pérdida de las proporciones corporales normales, que no siempre es fácilmente apreciada, dada la variabilidad normal que también existe en este aspecto. El examen de los progenitores puede en ocasiones ser de utilidad al poner de manifiesto deformidades no desarrolladas todavía en los niños. Las manifestaciones clínicas, los estudios de imagen y el patrón de herencia pueden orientar hacia una displasia ósea concreta o grupo de displasias (displasias espondilometafisarias, mesomélicas, acromélicas…) y dirigir los estudios moleculares a genes concretos o paneles de genes. Determinados genes, implicados en la regulación de la placa de crecimiento y responsables de diferentes formas de displasia ósea, han sido implicados en los últimos años con mayor frecuencia en casos de TBI, como por ejemplo: SHOX (short stature homeobox gen; Xp22.33 y Yp11.2), NPR2 (natriuretic peptide receptor B; 9p13), FGFR3 (fibroblast growth factor receptor 3; 4p16.3), NPPC (Natriuretic Peptide Precursor C; 2q37), ACAN (aggrecan; 15q26.1) y IHH (indian hedgehog homolog; 2q35). Muchos de estos genes (SHOX, NPR2, ACAN, etc.) en situaciones de homocigosis producen displasias óseas muy graves con marcada afectación de la talla y, en ocasiones, con grave disarmonía corporal; mientras que, en heterocigosidad serían responsables de formas menos extremas de talla baja armónica o mínimamente disarmónica susceptibles de ser etiquetadas de TBI.
Las manifestaciones clínicas, los estudios de imagen y el patrón de herencia pueden orientar hacia una displasia ósea concreta o grupo de displasias, y dirigir los estudios moleculares a genes concretos o paneles de genes
El desarrollo y difusión de nuevas técnicas de diagnóstico genético, especialmente los CGH-array (array-comparative genomic hybridization) y de secuenciación de nueva generación (paneles de genes, exoma o genoma) es lo que ha permitido avanzar en el diagnóstico de las causas monogénicas o genéticas de talla baja (cuadros dismórficos, displasias óseas, anomalías en los ejes hormonales, etc). Los estudios de CGH array ha demostrado que entre un 4 y un 10% de las tallas bajas podrían ser debidas a variaciones en el número de copias (CNVs), así como que la homocigosidad es un factor de riesgo de talla baja. Algoritmos de cómo aproximarse, desde un punto de vista diagnóstico, a las bases genéticas de la talla baja han sido propuestos recientemente(6) y es probable que, a medida que este tipo de estudios se abaraten y generalicen, pasen a formar parte de la evaluación habitual de niños con TBI, sobre todo de aquellos que asocien hallazgos que se ha demostrado que incrementan la posibilidad de encontrar una causa genética de su talla baja, como serían: anomalías congénitas o rasgos sindrómicos, datos sugerentes de displasia ósea, discapacidad intelectual, microcefalia, talla inferior a -3 SDS o predicción de talla por debajo de -2 SDS para la talla diana. El propósito de los estudios genéticos en el diagnóstico de la TBI radica no solo en llegar a obtener un diagnóstico que finalice la realización de pruebas diagnósticas, sino también a permitir realizar un seguimiento orientado a las comorbilidades asociadas al diagnóstico, realizar un adecuado consejo genético y valorar si una terapia está o no indicada o contraindicada, como podría ser, por ej. la contraindicación o prudencia del empleo de la GH en síndromes con predisposición al desarrollo de neoplasias (síndrome de Bloom, síndrome de Fanconi…).
Tratamiento de la TBI
Una vez establecido el diagnóstico de TBI, no existen criterios internacionales que establezcan qué pacientes deberían ser tratados ni cuál es el tratamiento más idóneo, aunque la rhGH y fármacos que modulan el inicio o el desarrollo de la pubertad han sido los más empleados(1,4,10,11).
Habitualmente, la decisión de tratar o no tratar suele basarse en aspectos auxológicos. En general, niños con talla inferior a -2 SDS, que están más de 2 SDS por debajo de su talla diana y cuya expectativa de talla se sitúa por debajo de -2 SDS para su edad y sexo serían los principales potenciales candidatos a tratamiento.
Los niños con talla inferior a -2 SDS, que están más de 2 SDS por debajo de su talla diana y cuya expectativa de talla se sitúa por debajo de -2 SDS para su edad y sexo serían los principales potenciales candidatos a tratamiento
Uno de los mayores problemas metodológicos a la hora de decidir la conveniencia o no de instaurar un tratamiento en pacientes con TBI es la escasa fiabilidad de los métodos de predicción de talla adulta basados en la EO del paciente. El método más utilizado para la valoración de la EO (método de Greulich-Pyle) muestra un error estándar de 0,55 a 0,82 años entre examinadores teóricamente entrenados. En lo referente al método de predicción de talla adulta más utilizado en la práctica clínica, el método de Bayley-Pinneau, que se basa en la EO alcanzada (diferencia tres grupos en cada sexo: normal, retrasada o adelantada) y el sexo, con frecuencia sobreestima la talla adulta en los casos de RCCP, especialmente si la EO está retrasada más de 2 años e infraestima la talla adulta en FSS, especialmente en niños.
El costo y duración del tratamiento, las molestias para el paciente y por supuesto los potenciales riesgos de “tratar” o “no tratar” al paciente son factores a tener en consideración a la hora de indicar un tratamiento en un niño, teóricamente sano, con TBI.
Dos de las premisas básicas que sostienen el empleo de tratamiento en la TBI, como son: que la talla baja supone un hándicap en la vida de un sujeto que requiere un adecuado tratamiento y que el aumento de la talla inducido por el tratamiento mejora la calidad de vida, no están plenamente demostradas. Aunque es claro que una talla baja extrema puede considerarse un hándicap, no hay clara evidencia de que tener una talla baja leve-moderada pueda ser considerado de la misma manera. Múltiples estudios han analizado desde el punto de vista psicológico las consecuencias en la infancia de una talla baja con/sin retraso puberal e incluso de las consecuencias de una talla baja en la edad adulta, pero los resultados de esas investigaciones no son concluyentes(10). En general, los niños con talla baja son objeto frecuente de bromas por sus compañeros e infantilizados por los adultos, lo que puede tener efectos negativos sobre su autoestima, ajuste social y conducta, pero la variabilidad individual es muy grande; de forma que, en algunos casos es posible demostrar una psicopatología asociada a la talla baja y en otros casos no. En adultos con talla baja, Christensen et al.(12) encontraron una a correlación significativa entre talla y calidad de vida; de forma que, los individuos con talla inferior a -2 SDS exhibían una reducción significativa en su calidad de vida. Es posible que un incremento de la talla inducido por el tratamiento pueda mejorar estas circunstancia, pero los beneficios del tratamiento, discretos en la mayoría de los casos, condicionan que muchos pacientes al finalizar el tratamiento continúen presentando una talla baja.
Testosterona y estradiol
En varones con TBI y retraso puberal (CDGP), este puede ser psicológicamente más importante que la talla baja que, además en muchos casos terminará situándose dentro de la normalidad. En estos casos, en que no es necesario incrementar la talla final, se han empleado desde hace años ciclos cortos de ésteres de testosterona, 3-6 meses, a dosis bajas (50-100 mg/mes por vía i.m) y a partir de los 13-14 años (EO >12 años). Estos ciclos aceleran el ritmo de crecimiento, el desarrollo de los caracteres sexuales secundarios y el propio inicio espontáneo de la pubertad sin comprometer la talla adulta. De igual forma, aunque menos frecuente, en niñas, el empleo de estrógenos a dosis bajas durante un periodo de 3-6 meses provoca similares efectos(10,11).
En varones con TBI y retraso puberal, se han empleado desde hace años ciclos cortos de ésteres de testosterona, de 3-6 meses, a dosis bajas y a partir de los 13-14 años
Análogos de GnRH
Los análogos de GnRH depot (triptorelina y leuprolide) son potentes agonistas de la GnRH (hormona hipotalámica liberadora de gonadotropinas) que probablemente por un mecismo de down-regulation de sus receptores inhiben la secreción de gonadotropinas y, secundariamente, de esteroides sexuales. Han sido utilizados desde los años ochenta y son el tratamiento estándar de la pubertad precoz central. La utilización de los análogos de GnRH (aGnRH) en pacientes con TBI con edad de inicio puberal normal, o en niñas con pubertad adelantada (inicio entre los 8 y 9 años) en un intento de alargar el tiempo de crecimiento y mejorar la talla adulta no se ha mostrado eficaz(13). Durante el tratamiento con aGnRH, la densidad mineral ósea disminuye y el IMC tiende a aumentar, aunque ambos fenómenos se normalizan tras la supresión del tratamiento.
Inhibidores de la aromatasa (IA)
La base para la utilización de estos fármacos radica en que varones con mutaciones inactivantes en el receptor de estrógenos o en la aromatasa, la enzima encargada de la síntesis de estrógenos a partir de precursores androgénicos, exceden su talla genética como consecuencia de un cierre tardío de las placas de crecimiento. Por tanto, la inhibición de la síntesis de estrógenos, mediante IA, podría potencialmente retrasar el cierre de las placas de crecimiento e incrementar la talla final en pacientes con TBI. La disminución de formación de estrógenos circulantes incrementa las gonadotropinas, lo que determina un incremento de los niveles de testosterona en varones y una hiperestimulación ovárica en niñas, motivo por el que estos fármacos no se utilizan en la TBI en niñas.
Los estudios iniciales en varones con RCCP y TBI, tratados con IA por vía oral (anastrozole o letrozole), mostraron clara mejoría en las predicciones de talla adulta (5-7 cm), pero los resultados a talla final, aunque muy escasos, no parecen reflejar el potencial beneficio sugerido. Una reciente revisión sistemática de Cochrane(14) ha puesto de manifiesto una escasa calidad en la evidencia de los estudios con IA en TBI, que no sostienen su utilización habitual. Además se han descrito potenciales efectos secundarios, entre ellos: aumento de deformidades vertebrales, marcado incremento de los niveles de testosterona, sobre todo en los tratados con letrozol, aumento de LDL-colesterol y triglicéridos y posible afectación de la espermatogénesis. En la actualidad, se considera que este tratamiento debería estar limitado a ensayos bien controlados(11).
Una reciente revisión sistemática ha puesto de manifiesto una escasa calidad en la evidencia de los estudios con IA en TBI, que no sostienen su utilización habitual
Hormona de crecimiento
El tratamiento con rhGH es el principal tratamiento empleado en la TBI, solo o asociado a moduladores de la pubertad. El primer ensayo de tratamiento con GH, durante 6 meses, en pacientes no deficientes de GH se realizó en 1983, todavía con pit-hGH y con aparentes resultados positivos (mejoría de 2 cm en la velocidad de crecimiento/anual); sin embargo y pese a los años transcurridos y múltiples ensayos clínicos llevados a cabo, todavía es controvertido el grado de efectividad de esta terapia en niños con TBI.
Un primer metanálisis con datos muy limitados (Finkelstein et al. en 2002)(15) sugirió una ganancia media de talla de 4-6 cm (rango de 2.3-8,7 cm), con una media de 1 cm por año de tratamiento. En 2011, Deodati y Cianfarini(16), llevaron a cabo un metaanálisis más detallado de los ensayos disponibles, incluyendo grados de calidad (alta, moderada, baja y muy baja) de acuerdo a la Sociedad de Endocrinología.
El objetivo era determinar de forma sistemática el impacto del tratamiento con GH en la talla adulta de niños con TBI. La revisión sistemática de la literatura mostró que de los 19 ensayos a largo plazo, sólo 10 eran ensayos controlados: 3 randomizados (RCTs), que incluían 115 niños (79 casos y 36 controles); y 7 no randomizados (non-RCTs), que incluían 477 niños (181 casos y 296 controles). De estos 10 ensayos, ninguno tenia una evidencia de alta calidad, dos RCTs fueron considerados de moderada calidad, 1 de baja-moderada calidad y 6 nonRCT de baja calidad. La mejoría media alcanzada en estos estudios fue de unos 4 cm (0,65 SDS) respecto a los controles, ligeramente inferior a la obtenida en otras situaciones de indicación de la rhGH. Por el contrario un reciente estudio “retrospectivo” en 123 niños con TBI y tratamiento con rhGH a dosis altas (0,046 mg/kg/día) sugiere beneficios superiores a 4 cm, con una media en varones de 9,5 cm (7,4-11,6 cm) y en mujeres de 8,6 cm (6,7-10,5 cm).
Salvo algunos estudios, como el ya comentado, la mayoría de ellos indican una mejoría media en la talla adulta de 4-5 cm (tras 4-5 años de tratamiento), pero con una significativa variabilidad interindividual en la respuesta a GH(18). No hay claros predictores de dicha respuesta a la GH y los pacientes se comportan, en este sentido, como un continuum. Se han sugerido como potenciales predictores de la respuesta a GH: un mayor déficit de talla respecto a la talla diana, un inicio del tratamiento más temprano (<10 años en niños y <9 años en niñas), así como una mayor elevación en los niveles de IGF-I. Padres más altos, mayor retraso en la EO y mejor predicción de talla al inicio del tratamiento se correlacionaron con mejores resultados de talla final. Hay también una clara relación dosis-respuesta, pero con gran variabilidad interpaciente; por lo que, se recomienda iniciar el tratamiento con la dosis más baja e incrementarla hasta que sea eficaz (entre 0,033 y 0,067 mg/kg/día). No hay datos que sostengan la utilización de dosis mayores de 0,067 mg/kg/día.
Recientemente, en 2016, la Sociedad de Endocrinología Pediátrica ha publicado una nueva guía para la utilización de la GH y el IGF-I en diferentes situaciones y, en concreto, en el caso de la TBI(19). Esta nueva guía, es la primera creada mediante el método GRADE (Grading of Recommendations Assessment, Development and Evaluation), un sistema de clasificación de la calidad de la evidencia y también un enfoque sistemático y transparente para el proceso de desarrollo de recomendaciones para la práctica clínica. Las recomendaciones de esta guía en lo referente a la TBI son poco específicas y más conservadoras que sus predecesoras:
La Sociedad de Endocrinología Pediátrica sugiere que la indicación de tratamiento con rhGH en niños con TBI debería realizarse solo “caso a caso”, después de analizar las repercusiones físicas y psicológicas y la ponderación de los riesgos-beneficios del tratamiento
- Sugieren que la indicación de tratamiento con rhGH en niños con TBI debería realizarse solo “caso a caso”, después de analizar las repercusiones físicas y psicológicas y la ponderación de los riesgos-beneficios del tratamiento. No se recomienda el tratamiento rutinario en niños con talla < -2,25 SDS (calidad de la evidencia: moderada). Aunque los tratamientos con rhGH han demostrado un incremento de la talla media en las cohortes de pacientes con TBI tratados con GH, hay una marcada variabilidad en la respuesta, incluyendo algunos individuos que no responden a tratamiento.
- Sugieren que debería realizarse una valoración de los beneficios obtenidos en lo referente a SDS de talla e impacto psicológico al cabo de un año de tratamiento y optimización de dosis (calidad de la recomendación: baja).
- Debido al solapamiento en las respuestas entre diferentes dosis, la guía sugiere iniciar el tratamiento con rhGH a la dosis de 0,035 mg/kg/día, aunque algunos pacientes requerirán aumentarla a 0,067 mg/kg/día (calidad de la recomendación: baja).
- Se recomienda que la evaluación y tratamiento de estos niños la realicen médicos con experiencia (norma de buena práctica).
La nueva guía recomienda valorar la respuesta al tratamiento al cabo de un año, sin embargo no especifica cómo realizar dicha valoración. La criterios auxológicos empleados para valorar la respuesta a la rHGH son variables en los diferentes estudios(20) y ni la velocidad de crecimiento, ni el cambio en SDS de talla ni la mejoría en la predicción de talla adulta predicen de manera fiable la mejoría en la talla adulta.
La combinación de rhGH y análogos de GnRH en el tratamiento de la TBI ha sido ampliamente utilizada, pero los datos disponibles solo sugieren una mejoría discreta(13). Un ensayo randomizado y controlado(21), para analizar los efectos sobre la talla adulta de una combinación de rhGH con aGnRH vs no tratamiento, en 32 adolescentes con TBI, expectativa de talla adulta inferior a -2 SDS y pubertad relativamente temprana (estadio II-III de Tanner), produjo un beneficio medio en la talla adulta de los pacientes tratados de solo 4,9 cm, aunque con gran variabilidad interindividual (rango entre -4 cm y +12,3 cm).
La combinación de rhGH con IA(22) o con IGF-1(23) en el tratamiento de la TBI, aunque parece inicialmente prometedora, requiere de mayores estudios y no debería aplicarse salvo en ensayos controlados.
La seguridad de la GH en el tratamiento de la talla baja ha sido estudiada a lo largo de más de 30 años. En concreto, en los niños con TBI, los efectos secundarios de la rhGH son escasos y similares, incluso menores, a los observados en otras formas de talla baja tratadas con rhGH (hipertensión intracraneal, deslizamiento de la cabeza femoral, rasgos acromegaloides, escoliosis, pancreatitis…)(24,25); no obstante, su seguridad a largo plazo ha sido cuestionada recientemente por estudios observacionales que sugerían un riesgo aumentado de mortalidad y morbilidad en adultos jóvenes tratados con GH durante la infancia(26,27). Aunque estos datos no han sido confirmados, no deja de existir un cierto grado de incertidumbre sobre sus potenciales efectos secundarios a largo plazo; por lo que, parece esencial continuar el seguimiento de los pacientes tratados, más aún cuando la indicación para el uso de la GH continúa siendo cuestionable.
Bibliografía
- Deodati A, Cianfarani S. The Rationale for Growth Hormone Therapy in Children with Short Stature. J Clin Res Pediatr Endocrinol. 2017; 9 (suppl2): 23-32.
- Ranke MB. Towards a consensus on the definition of idiopathic short stature. Horm Res. 1996; 45 (suppl 2): 64-66.
- Wit JM, Clayton PE, Rogol AD, Savage MO, Saenger PH, Cohen P. Idiopathic short stature: definition, epidemiology, and diagnostic evaluation. Growth Horm IGF Res. 2008;18: 89-110.
- Wit JM, Reiter EO, Ross JL, Saenger PH, Savage MO, Rogol AD, Cohen P. Idiopathic short stature: management and growth hormone treatment. Growth Horm IGF Res. 2008;18: 111-35.
- Cohen P, Rogol AD, Deal CL, Saenger P, Reiter EO, Ross JL, Chernausek SD, Savage MO, Wit JM; 2007 ISS Consensus Workshop participants. Consensus statement on the diagnosis and treatment of children with idiopathic short stature: a summary of the Growth Hormone Research Society, the Lawson Wilkins Pediatric Endocrine Society, and the European Society for Paediatric Endocrinology Workshop. J Clin Endocrinol Metab. 2008; 93: 4210-4217.
- Dauber A, Rosenfeld RG, Hirschhorn JN. Genetic evaluation of short stature. J Clin Endocrinol Metab. 2014;99: 3080-3092.
- Díaz-López I, Poncela-García JA, Carranza-Ferrer M. Uso fuera de ficha clínica de la hormona de crceimineto. Aspectos legislativos y revisión científica de su evidencia. Rev Esp Endocrinol Pediatr 2017; 8: 30-41.
- Wit JM, Oostdijk W, Losekoot M, van Duyvenvoorde HA, Ruivenkamp CA, Kant SG. MECHANISMS IN ENDOCRINOLOGY: Novel genetic causes of short stature. Eur J Endocrinol. 2016; 174: R145-73.
- Argente J, Pérez-Jurado LA. Genetic causes of proportionate short stature. Best Pract Res Clin Endocrinol Metab. 2018; 32: 499-522.
- Ranke MB. Treatment of children and adolescents with idiopathic short stature. Nat Rev Endocrinol. 2013; 9: 325-334.
- Wit JM, Oostdijk W. Novel approaches to short stature therapy. Best Pract Res Clin Endocrinol Metab. 2015; 29: 353-366.
- Christensen TL, Djurhuus CB, Clayton P, Christiansen JS. An evaluation of the relationship between adult height and health-related quality of life in the general UK population. Clin Endocrinol (Oxf). 2007; 67: 407-412.
- Carel JC. Management of short stature with GnRH agonist and co-treatment with growth hormone: a controversial issue. Mol Cell Endocrinol. 2006; 254-255: 226-33.
- McGrath N, O’Grady MJ. Aromatase inhibitors for short stature in male children and adolescents. Cochrane Database of Systematic Reviews 2015, Issue 10. Art. No.: CD010888. DOI: 10.1002/14651858.CD010888.pub2.
- Finkelstein BS, Imperiale TF, Speroff T, Marrero U, Radcliffe DJ, Cuttler L. Effect of growth hormone therapy on height in children with idiopathic short stature: a meta-analysis. Arch Pediatr Adolesc Med. 2002;156: 230-240.
- Deodati A, Cianfarani S. Impact of growth hormone therapy on adult height of children with idiopathic short stature: systematic review. BMJ. 2011; 342:c7157. doi: 10.1136/bmj.c7157.
- Sotos JF, Tokar NJ. Growth hormone significantly increases the adult height of children with idiopathic short stature: comparison of subgroups and benefit. Int J Pediatr Endocrinol. 2014; 1:15. doi: 10.1186/1687-9856-2014-15.
- Bryant, J, Baxter, L, Cave, CB, Milne, R. Recombinant growth hormone for idiopathic short stature in children and adolescents. Cochrane Database of Systematic Reviews, Issue 3. Art. No.: CD004440. http://dx.doi.org/10.1002/14651858.CD004440.pub2
- Grimberg A, DiVall SA, Polychronakos C, Allen DB, Cohen LE, Quintos JB, Rossi WC, Feudtner C, Murad MH; Drug and Therapeutics Committee and Ethics Committee of the Pediatric Endocrine Society. Guidelines for Growth Hormone and Insulin-Like Growth Factor-I Treatment in Children and Adolescents: Growth Hormone Deficiency, Idiopathic Short Stature, and Primary Insulin-Like Growth Factor-I Deficiency. Horm Res Paediatr. 2016; 86: 361-397.
- Bang P, Bjerknes R, Dahlgren J, Dunkel L, Gustafsson J, Juul A, Kristrom B, Tapanainen P, Aberg V: A comparison of different definitions of growth response in short prepubertal children treated with growth hormone. Horm Res Paediatr 2011; 75: 335–345.
- van Gool SA1, Kamp GA, Visser-van Balen H, Mul D, Waelkens JJ, Jansen M, et al. Final height outcome after three years of growth hormone and gonadotropin-releasing hormone agonist treatment in short adolescents with relatively early puberty. J Clin Endocrinol Metab. 2007;92: 1402-1408.
- Mauras N, Ross JL, Gagliardi P, Yu YM, Hossain J, Permuy J, et al. Randomized Trial of Aromatase Inhibitors, Growth Hormone, or Combination in Pubertal Boys with Idiopathic, Short Stature. J Clin Endocrinol Metab. 2016;101: 4984-4993.
- Backeljauw PF, Miller BS, Dutailly P, Houchard A, Lawson E, Hale DE, Reiner B, Sperling MA; MS316 Study Group. Recombinant human growth hormone plus recombinant human insulin-like growth factor-1 coadministration therapy in short children with low insulin-like growth factor-1 and growth hormone sufficiency: results from a randomized, multicenter, open-label, parallel-group, active treatment-controlled trial. Horm Res Paediatr. 2015; 83: 268-279.
- Allen DB. Safety of growth hormone treatment of children with idiopathic short stature: the US experience. Horm Res Paediatr. 2011;76 Suppl 3:45-47.
- Bell J, Parker KL, Swinford RD, Hoffman AR, Maneatis T, Lippe B. Long-term safety of recombinant human growth hormone in children. J Clin Endocrinol Metab. 2010; 95: 167-177.
- Carel JC, Ecosse E, Landier F, Meguellati-Hakkas D, Kaguelidou F, Rey G, Coste J. Long-term mortality after recombinant growth hormone treatment for isolated growth hormone deficiency or childhood short stature: preliminary report of the French SAGhE study. J Clin Endocrinol Metab 2012; 97: 416-425.
- Poidvin A, Touzé E, Ecosse E, Landier F, Béjot Y, Giroud M, Rothwell PM, Carel JC, Coste J. Growth hormone treatment for childhood short stature and risk of stroke in early adulthood. Neurology 2014;83:780-786.